ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2021; 9:20-25. doi:10.7150/jgen.53678 This volume Cite
Research Paper
Draft genome sequences of three filamentous cyanobacteria isolated from brackish habitats
Joanne Sarah Boden1 ![]() , Michele Grego2, Henk Bolhuis2, Patricia Sánchez-Baracaldo1
, Michele Grego2, Henk Bolhuis2, Patricia Sánchez-Baracaldo1 ![]()
1. School of Geographical Sciences, Faculty of Science, University of Bristol, Bristol, BS8 1SS, United Kingdom.
2. Department of Marine Microbiology and Biogeochemistry, Royal Netherlands Institute for Sea Research, and Utrecht University, Den Hoorn, the Netherlands.
Received 2020-9-24; Accepted 2021-1-27; Published 2021-2-17
Abstract

Brackish cyanobacterial genome sequences are relatively rare. Here, we report the 5.5 Mbp, 5.8 Mbp and 6.1 Mbp draft genomes of Spirulina sp. CCY15215, Leptolyngbya sp. CCY15150 and Halomicronema sp. CCY15110 isolated from coastal microbial mats on the North Sea beach of the island of Schiermonnikoog in the Netherlands. Large scale phylogenomic analyses reveal that Spirulina sp. CCY15215 is a large cell diameter cyanobacterium, whereas Leptolyngbya sp. CCY15150 and Halomicronema sp. CCY15110 are the first reported brackish genomes belonging to the LPP clade consisting primarily of Leptolyngbya, Plectonema and Phormidium spp. Further genome mining divulges that all new draft genomes contain, ggpS and ggpP, the genes responsible for synthesising glucosylglycerol (GG), a compatible solute found in moderately salt-tolerant cyanobacteria.
Keywords: Spirulina sp. CCY15215, Halomicronema sp. CCY15110, Leptolyngbya sp. CCY15150, cyanobacteria, genome, brackish
Introduction
Cyanobacteria (commonly known as 'blue-green algae') are oxygenic photoautotrophs which evolved in the Archaean (4 to ~2.5 billion years ago) [1, 2] and have since diversified into a wide range of habitats, including lakes, oceans, soil, rocks, geothermal springs and glaciers [3]. Brackish species have been found living in estuarine environments [4, 5], but few genomes of brackish species have been sequenced [6]. Some of these include Nodularia spumigena CCY9414 and Aphanizomenon flos-aquae from the Baltic Sea [7, 8] and four strains of Microcystis aeruginosa from Japan and the Netherlands [9-11].
Phylogenetic and trait evolution studies suggest there have been several transitions between saltwater and freshwater environments during the evolution of cyanobacteria [12]. Intermediate stages of such transitions could have lived in brackish environments before acquiring the genetic mechanisms (e.g. synthesis of compatible solutes [13]) necessary to tolerate higher salinity of the ocean or lower salinity of freshwater lakes and rivers inland. To understand these stages in more detail, we sequenced draft genomes of three new brackish and filamentous strains isolated from sediments on the island of Schiermonnikoog in The Netherlands: Spirulina sp. CCY15215, Halomicronema sp. CCY15110 and Leptolyngbya sp. CCY15150.
Materials and Methods
Three strains of cyanobacteria were isolated from a microbial mat on the island of Schiermonnikoog in The Netherlands [14]. Mono-phototrophic cultures were isolated by plating and grown in BA+ medium (https://www.ccy.nioz.nl/cyanobacteria_media) at 20°C under a 16 h: 8 h light:dark cycle with 10-20 μmol m-2 s-1 of white light.
Genomic DNA was extracted from 1.8 ml of each microbial culture using DNeasy ultraclean microbial kits (Qiagen, Germany) according to the manufacturer's instructions. Purified genomic DNA was stored in 10 mM Tris buffer at pH 8 and -80°C until further use.
Genomic DNA libraries for each strain were prepared using the TruSeq Nano LT kit (Illumina, Inc, Cambridge, UK). Whole genome sequencing was performed by the University of Bristol Genomics Facility, UK. Paired-end reads of 2x150 bps were obtained using a NextSeq® 500/550 (Illumina, San Diego, CA) with 300 cycles of the Mid Output Kit v2. Raw reads were quality trimmed using Trimmomatic v0.39 [15] and de novo assembly performed with SPAdes v3.14.0 [16]. Short contigs containing less than 200 nucleotides were removed from the resulting assemblies. Cyanobacterial contigs were separated from other contigs using BLAST analysis and a de Bruijn graph visualisation approach that has previously been used to assemble draft genomes of Phormidesmis priestleyi BC1401 [17] and Leptolyngbya sp. BC1307 [18]; see Supplementary Information Figures S1-S3. Genome completeness was measured based on comparisons with single-copy orthologs from cyanobacteria using BUSCO v3.0.2 [19]. All draft genomes were submitted to JGI IMG/ER [20] for annotation (GOLD Analysis Project IDs: Ga0438272, Ga0438271 and Ga0437567). Draft genome sequences have been deposited in DDBJ/ENA/Genbank repositories under accession numbers JACSWA000000000 for Spirulina sp. CCY15215, JACSWB000000000 for Leptolyngbya sp. CCY15150 and JACSWC000000000 for Halomicronema sp. CCY15110. These are stored within BioProject PRJNA658956 with BioSample accessions from SAMN15893520 to SAMN15893522.
The evolutionary relationships of our brackish strains with other cyanobacteria were estimated by performing maximum likelihood phylogenetic analysis of 139 orthologous proteins, SSU rRNA and LSU rRNA from 168 strains representing the entire diversity of the Phylum, Cyanobacteria. Similar methods have been implemented previously (see these studies for a more detailed description [6, 12, 21]). Briefly, genomes were obtained from the NCBI RefSeq database (https://www.ncbi.nlm.nih.gov/refseq/). Protein and rRNA sequences were identified using the basic local alignment search tool (BLAST) for proteins or nucleotides respectively with an e value cut off <1×10-25 to remove anomalous hits. Each protein and rRNA were aligned using MAFFT v7.427 [22] and gaps removed if present in 85% or more sequences. The phylogeny itself was constructed in IQ-TREE v1.6.7 [23] using partitioned analyses which account for heterotachy and allow each protein and rRNA to evolve under a substitution model appropriate to that unique protein or RNA. Each of the substitution models were selected using Bayesian Information Criterion scores predicted by ModelFinder [24]. Support for branching relationships were measured by calculating ultrafast bootstrap approximations with 1000 replicates [25]. The resulting phylogeny was rooted with four representative strains of Vampirovibrionia, the sister phylum of Cyanobacteria [26].
Results and Discussion
Each draft genome is composed from between 304 and 621 contigs with genome sizes ranging from 5.5 to 6.1 Mbps. Overall genome completeness is estimated at between 97.1 to 98.0% with N50 values in the range of 31,079 to 116,290 and GC content from 42.6 to 54.6% (Table 1). To find out how these strains are related to other cyanobacteria, phylogenomic analyses were conducted using 139 orthologous cyanobacterial proteins as well as SSU and LSU rRNA. They reveal that 2 of the 3 filamentous brackish strains are nested within the LPP clade previously defined by Sánchez-Baracaldo [6] (Figure 1, Supplementary Information Figure S4). The other, named Spirulina sp. CCY15215 is in a distant phylogenetic position within the Macrocyanobacterial clade defined by Sánchez-Baracaldo [12] (Figure 1). Macrocyanobacteria are ecologically diverse, but generally have large cells, ranging from 3 to 50 μm in diameter. Members of the clade vary from Nostocales to Microcystis spp. and Oscillatoria spp.
Genome Features
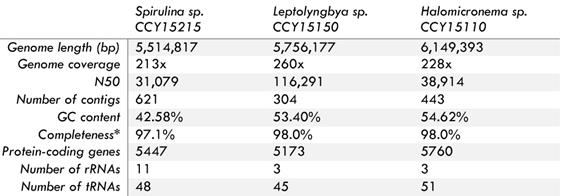
*Completeness was estimated using BUSCO v3.0.2 with lineage data from cyanobacteria [19].
Phylogenetic tree showing the relationships of Spirulina sp. CCY15215, Halomicronema sp. CCY15110 and Leptolyngbya sp. CCY15150 (red) within the Phylum, Cyanobacteria. The tree was constructed from alignments of 139 orthologous proteins, SSU rRNA and LSU rRNA using Maximum Likelihood methodology implemented in IQ-TREE v1.6.7 [23]. It was rooted using four strains of Vampirovibrionia as the outgroup. Node labels represent ultrafast bootstrap approximations with circles representing values of 100. The scale bar represents an average of 0.2 substitutions per site. For a complete tree detailing all taxa that were included, see Supplementary Information Figure S4.
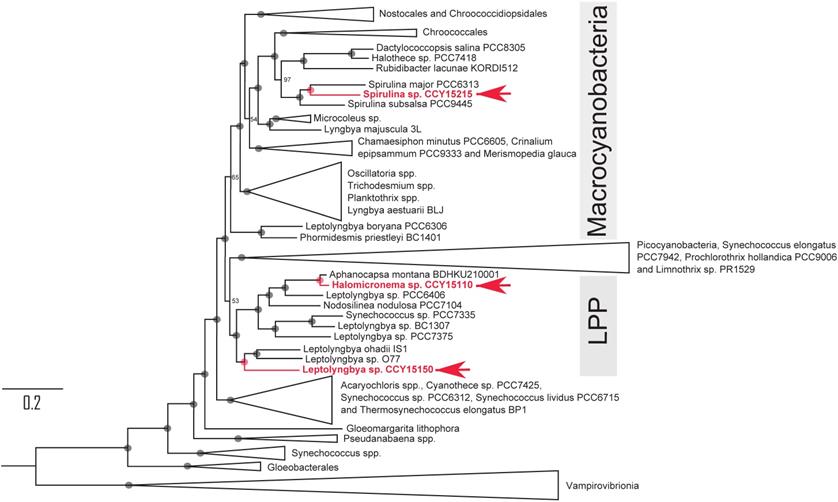
In particular, Spirulina sp. CCY15215 shares a recent common ancestor with two other Spirulina spp. that have slightly smaller genome sizes of 5.05 and 5.32 Mbp [27] compared to 5.5 Mbp for Spirulina sp. CCY15215. The closest relative, Spirulina major PCC6313 was also isolated from brackish waters [27]. It was found in California, North America [27], more than 8000 km away from the Netherlands where Spirulina sp. CCY15215 was collected from. Their outgroup, Spirulina subsalsa PCC9445, has previously been cultured in artificial seawater [28]. Since these close relatives of Spirulina sp. CCY15215 also grow in brackish and saltwater habitats, the most recent common ancestor of Spirulina spp. may also have been salt-tolerant.
Unlike Spirulina sp. CCY15215 which shares a common ancestor with brackish cyanobacteria, Halomicronema sp. CCY15110 is sister to a marine strain, named Aphanocapsa montana BDHKU 210001 (Figure 1). Phylogenetic analyses conducted using 16S SSU rRNA show that Halomicronema sp. CCY15110 is also closely-related to Halomicronema excentricum str. Lakshadweep, isolated from a marine habitat in India (Supplementary Information, Figure S5). The genus name 'Halomicronema' was therefore assigned to reflect this relationship.
To further investigate the mechanisms responsible for salt tolerance, protein BlastP searches were conducted for enzymes involved in the production of compatible solutes. Compatible solutes are molecules which aid osmoregulation in salty water [29]. Their production is catalysed by a series of genome-encoded enzymes, including phosphatases (eg. GpgP, GgpP, Spp), synthases (eg. GpgS, GgpS, TreY, Sps), hydrolases (eg. TreZ) and methyltransferases (eg. GSMT and DMT). In order to make any single compatible solute, multiple enzymes are required. For example, an appropriate synthase and phosphatase must be present to combine the glucose group of one molecule with the carbohydrate group of another and then remove a phosphate ion to synthesise sucrose, glucosylglycerol (GG) or glucosylglycerate (GGA). Whereas, to synthesise trehalose and glycine betaine (GB), either a synthase and hydrolase or two different methyltransferases must be encoded respectively in the genome [29]. To ensure we correctly identified the presence or absence of genes encoding these enzymes in each new genome, BlastP were conducted with query sequences from well-characterised cyanobacteria, including Synechocystis sp. PCC 6803, Nostoc sp. PCC 7120 and Synechococcus sp. PCC 7002 amongst others (Supplementary Information, Table S1).
Our genome mining reveals that all three brackish strains are genetically capable of making GG (Table 2). It has previously been hypothesised that GG-production is a characteristic of moderately halotolerant marine cyanobacteria [29]. In addition to GG, Leptolyngbya sp. CCY15150 and Halomicronema sp. CCY15110 can make GGA (Table 2). Experimental studies measuring the accumulation of compatible solutes in cyanobacteria exposed to changing environmental conditions, suggest that GGA production increases in response to nitrogen limitation as well as increasing salinity [30], so its presence in Leptolyngbya sp. CCY15150 and Halomicronema sp. CCY15110 may help them to grow in lower nitrogen concentrations.
Results of BLASTP for genes encoding compatible solutes in three new draft genomes of cyanobacteria isolated from an overall brackish habitat
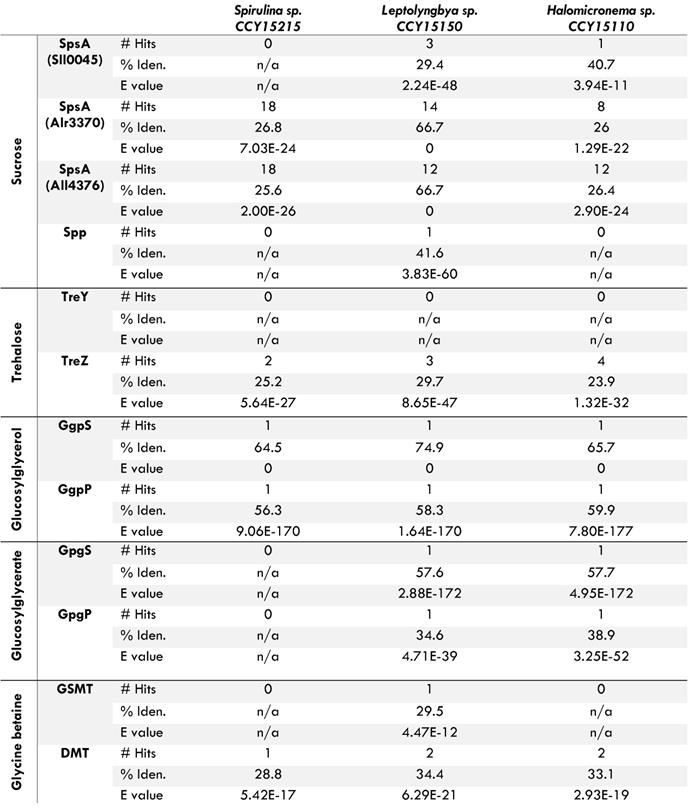
Homologs of genes required for the biosynthesis of sucrose (SpsA and Spp), trehalose (TreY and TreZ), glucosylglycerol (GgpS and GgpP), glucosylglycerate (GpgS and GpgP) and glycine betaine (GSMT and DMT) were found via BLASTP for genes identified by [29]. All results have e values lower than 1x10-10. If multiple hits were found, statistics are reported for the hit with the smallest e value. % Iden. refers to percent identity.
In addition to GG and GGA, Leptolyngbya sp. CCY15150 is genetically capable of making two further compatible solutes which may help it tolerate more saline conditions on the coastline. These are GB and sucrose (Table 2). Similarly large ranges of compatible solutes can be found in hypersaline strains [13], which is unusual given that Leptolyngbya sp. CCY15150 was isolated from a fresher, brackish environment [14]. One explanation may lie in the changeability of intertidal habitats. Coastal microbial mats are subjected to alternate ebbs and flows of the tide and changing weather patterns (e.g. precipitation, humidity and temperature) which have the effect of changing salinity. For example, on hot days, after the tide recedes, water evaporates from the microbial mat, leaving the surrounding environment to become more and more saline until the next rainfall or influx of the tide. Metagenomic analyses show that during Summer, the hottest season of the year in Schiermonnikoog, Leptolyngbya spp. in general are more abundant than Halomicronema spp. [14]. The presence of additional compatible solutes in Leptolyngbya sp. CCY15150 may go some way towards explaining this. Conversely, as draft genomes of Halomicronema sp. CCY15110 and Spirulina sp. CCY15215 both contain homologs of genes encoding one of two enzymes required for sucrose biosynthesis (SpsA but not Spp) and one of two enzymes required for GB biosynthesis (DMT but not GSMT, Table 2), there remains a small possibility that their complete genomes are sufficient to make additional compatible solutes.
Overall, draft genomes of three filamentous cyanobacteria expand the genomic representation of brackish strains in the tree of life and reveal that strains isolated from the same intertidal environment employ different strategies to acclimate to and tolerate variable salinities in a changing environment.
Abbreviations
BLAST: basic local alignment search tool; GG: glucosylglycerol; GGA: glucosylglycerate; GB: glycine betaine; SSU: small sub-unit; rRNA: ribosomal ribonucleic acid; LSU: large sub-unit; RNA: ribonucleic acid.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Giorgio Bianchini for providing scripts to gather phylogenetic data sets, and Dan Macrae for assistance with DNA extraction and culture maintenance of Spirulina sp. CCY15215. Funding support for this work came from a Royal Society Research Fellows Enhancement Award (RGF\EA\180219) and a Royal Society University Research Fellowship to P.S-B.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schirrmeister BE, Gugger M, Donoghue PCJ. Cyanobacteria and the Great Oxidation Event: Evidence from genes and fossils. Palaeontology. 2015;58:769-85
2. Schirrmeister BE, Sánchez-Baracaldo P, Wacey D. Cyanobacterial evolution during the Precambrian. Int. J. Astrobiology. 2016;15:187-204
3. Mishra AK, Tiwari DN, Rai AN. Cyanobacteria: From basic science to applications. 1st ed. London, United Kingdom: Academic Press. 2018
4. Celepli N, Sundh J, Ekman M, Dupont CL, Yooseph S, Bergman B. et al. Meta-omic analyses of Baltic Sea cyanobacteria: Diversity, community structure and salt acclimation. Environ. Microbiol. 2017;19:673-86
5. Lopes VR, Vasconcelos VM. Planktonic and benthic cyanobacteria of European brackish waters: A perspective on estuaries and brackish seas. Eur. J. Phycol. 2011;46:292-304
6. Blank CE, Sánchez-Baracaldo P. Timing of morphological and ecological innovations in the cyanobacteria - a key to understanding the rise in atmospheric oxygen. Geobiology. 2010;8:1-23
7. Bolhuis H, Cretoiu MS, Stal LJ. Molecular ecology of microbial mats. FEMS Microbiol. Ecol. 2014;90:335-50
8. Sulcius S, Alzbutas G, Kvederaviciute K, Koreiviene J, Zakrys L, Lubys A. et al. Draft genome sequence of the cyanobacterium Aphanizomenon flos-aquae strain 2012/KM1/D3, isolated from the Curonian Lagoon (Baltic Sea). Genome Announc. 2015 3
9. Tanabe Y, Yamaguchi H. Draft genome sequence of Microcystis aeruginosa NIES-4285, isolated from brackish water (Lake Abashiri, Japan). Microbiol. Resour. Announc. 2019 8
10. Sandrini G, Huisman J, Matthijs HC. Potassium sensitivity differs among strains of the harmful cyanobacterium Microcystis and correlates with the presence of salt tolerance genes. FEMS Microbiol. Lett. 2015 362
11. Tanabe Y, Hodoki Y, Sano T, Tada K, Watanabe MM. Adaptation of the freshwater bloom-forming cyanobacterium Microcystis aeruginosa to brackish water is driven by recent horizontal transfer of sucrose genes. Front. Microbiol. 2018;9:1150
12. Sánchez-Baracaldo P. Origin of marine planktonic cyanobacteria. Sci. Rep. 2015;5:17418
13. Hagemann M. Molecular biology of cyanobacterial salt acclimation. FEMS Microbiol. Rev. 2011;35:87-123
14. Cardoso DC, Cretoiu MS, Stal LJ, Bolhuis H. Seasonal development of a coastal microbial mat. Sci. Rep. 2019;9:9035
15. Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20
16. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455-77
17. Chrismas NAM, Barker G, Anesio AM, Sánchez-Baracaldo P. Genomic mechanisms for cold tolerance and production of exopolysaccharides in the Arctic cyanobacterium Phormidesmis priestleyi BC1401. BMC Genomics. 2016;17:533
18. Chrismas NAM, Williamson CJ, Yallop ML, Anesio AM, Sánchez-Baracaldo P. Photoecology of the Antarctic cyanobacterium Leptolyngbya sp. BC1307 brought to light through community analysis, comparative genomics and in vitro photophysiology. Mol. Ecol. 2018;27:5279-93
19. Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210-2
20. Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y. et al. IMG: The integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40:D115-22
21. Sánchez-Baracaldo P, Bianchini G, Di Cesare A, Callieri C, Chrismas NAM. Insights Into the evolution of Picocyanobacteria and phycoerythrin genes (mpeBA and cpeBA). Front. Microbiol. 2019;10:45
22. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013;30:772-80
23. Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44:W232-5
24. Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587-589
25. Hoang DTC O. Haeseler,A.V. Minh,B.Q. Vinh,L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2017;35:518-22
26. Soo RM, Skennerton CT, Sekiguchi Y, Imelfort M, Paech SJ, Dennis PG. et al. An expanded genomic representation of the Phylum Cyanobacteria. Genome Biol. Evol. 2014;6:1031-45
27. Shih PM, Wu DY, Latifi A, Axen SD, Fewer DP, Talla E. et al. Improving the coverage of the Cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. USA. 2013;110:1053-8
28. Cohen Z, Vonshak A. Fatty acid composition of Spirulina and Spirulina-like cyanobacteria in relation to their chemotaxonomy. Phytochemistry. 1991;30:205-6
29. Hagemann M. Genomics of salt acclimation: Synthesis of compatible solutes among cyanobacteria. Adv. Bot. Res. 2013;65:27-55
30. Klahn S, Steglich C, Hess WR, Hagemann M. Glucosylglycerate: A secondary compatible solute common to marine cyanobacteria from nitrogen-poor environments. Environ. Microbiol. 2010;12:83-94
Author contact
![]() Corresponding authors: Joanne Sarah Boden and Patricia Sánchez-Baracaldo. E-mail(s): j.bodenac.uk; p.sanchez-baracaldoac.uk.
Corresponding authors: Joanne Sarah Boden and Patricia Sánchez-Baracaldo. E-mail(s): j.bodenac.uk; p.sanchez-baracaldoac.uk.