ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2020; 8:25-29. doi:10.7150/jgen.43162 This volume Cite
Research Paper
Draft Genome Analysis of Christensenella minuta DSM 22607, exhibiting an unusual expansion of transporter homologs of unknown function
David A Coil1, Guillaume Jospin1, Jonathan A. Eisen1,2,3,4 ![]()
1. Genome Center, University of California, Davis, CA, United States.
2. Department of Evolution and Ecology, University of California, Davis, CA, United States.
3. Center for Population Biology, University of California, Davis, California, United States.
4. Department of Medical Microbiology and Immunology, University of California, Davis, CA, United States.
Received 2019-12-17; Accepted 2020-2-10; Published 2020-2-21
Abstract

Christensenella minuta was first formally described in 2012 as a member of a novel species, genus, and proposed family of Christensenellaceae. C. minuta was later shown in one study to be part of the most heritable taxonomic group in the human gut microbiome and to be enriched in people with low body mass index (BMI). Mouse work demonstrated that injection of cultured C. minuta into germ-free mice prevented the onset of obesity after a fecal transplant to the mice from high BMI individuals. Here we describe the genome sequence of C. minuta DSM 22607. Examination and analysis of the annotation revealed an unusually high number of genes predicted to be involved in carbohydrate metabolism, many of which were multiple homologs of RbsA, RbsB and RbsC, which together make up the Ribose ABC Transport System. These genes may be also involved in quorum sensing which could potentially relate to the importance of C. minuta in the gut microbiome.
Keywords: Christensenella minuta, C. minuta DSM 22607, Draft Genome Analysis
Introduction
A strain of Christensenella minuta, isolated from human feces, was described in 2012 as a member of a novel species, genus, and proposed family of Christensenellaceae [1]. C. minuta and the Christensenellaceae were highlighted as potentially of significant interest in 2014 in a paper involving studies of human twins and also experiments in mice [2]. Findings from this paper include (a) that the Christensenellaceae family was the most heritable taxonomic group in the human gut microbiome, (b) that the Christensenellaceae and some other associated taxa were enriched in people with low body mass index (BMI) and (c) that injection of cultured C. minuta into germ-free mice prevented the onset of obesity after a fecal transplant to the mice from high BMI individuals. The mechanisms underlying these findings were not revealed in this study, and the story was further complicated by subsequent work demonstrating the pathogenic potential of C. minuta [3]. In the last few years, there have been a number of studies showing human health associations with Christensenellacaeae, ranging from serum lipid levels to longevity to metabolic disorders [4]. Based on the results of the 2014 paper and given the putative importance of C. minuta in human health, we sought to further examine C. minuta by sequencing its genome since none was publically available at the time. The genome of the type strain DSM 22607 has been since sequenced several times but the only published genome report was a data announcement [5]. We report here sequencing efforts and a brief analysis of the genome of C. minuta strain DSM 22607. Specifically, we highlight the phylogenetically recent massive expansion of a set of transporter genes of unclear function.
Methods
Genome Sequencing
DNA from C. minuta DSM 22607 was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). A paired-end library for Illumina sequencing was created using a Nextera XT Library Preparation Kit (Illumina). The library was size-selected (600-900 bp) on a Pippin Prep system (Sage Science) and sequenced on a PE300 bp run of an Illumina MiSeq at the UC Davis DNA Technologies Core Facility. Quality trimming, error correction, and assembly were performed by the A5-miseq assembly pipeline [6]. Genome annotation was examined using RAST and the associated SEED Viewer [7].
Whole genome tree and HMM scanning
Genomes of all 9499 sequenced type strains were downloaded from NCBI on August 23rd 2019. These were all run through Phylosift's [8] search (lastal [9]) and align (hmmalign [10]) functions to extract 37 mostly single copy marker genes used to build a large “whole genome” phylogenetic tree (FastTreeMP, [11]). From C. minuta's location on the tree, we walked back up to an internal node to include two large clades around C. minuta DSM 22607. Streptococcus porcinus was selected as belonging to a closely related clade to the three described previously. This resulted in 373 taxa being selected for a maximum likelihood tree using RAxML 8.2.11 [12] and the PROTGAMMABLOSUM62 substitution matrix, and the following parameters (-p 8 -x 47 -T 48 -f a -N 1000). Using the C. minuta DSM 22607 genome annotation from RAST, we extracted the sequences of all RbsA (n=42), RbsB (n=20), and RbsC (n=41) proteins. Each of these sequences was then compared to the progidal2.6.3 [13] predicted proteins from the 9,499 genomes using BLAST2.9 [14]. Predicted proteins matching the reference proteins below an e-value of 1e-30, were counted and summed for each genome and added to the tree's tip labels. Any hits below an e-value of 1e-30 were counted for each marker. These numbers were added to the tree's tip labels.
Average nucleotide identity (ANI)
We selected all 11 publicly available Christensenella genomes and compared their average nucleotide identity using fastANI [15] with default settings (fragment size of 3000bp).
Results & Discussion
Assembly/Validation
In total 770,216 Illumina reads were used in the assembly, producing a draft genome of 2,942,834 bp, in 42 contigs, with an N50 of 148,400 bp at a coverage of ~79X. Genome completeness was estimated using CheckM [16], which searches for single-copy, highly conserved markers, and it gave a completeness estimate of 98.4 % and contamination estimate of .81%.
Annotation
We sought to make predictions regarding the unique metabolic characteristics of C. minuta, but this was complicated by the lack of genome sequences for close relatives. According to the original species description of C. minuta, the closest relatives were Caldicoprobacter oshimai, Tindallia californiensis, and Clostridium ganghwense [1] (which aren't even from the same family within Clostridiales). Both C. oshimai and T. californiensis have sequenced genomes, and these were used here for an initial comparison. The RAST Seed Viewer also highlights the genome that the RAST automated analysis identifies as having the most similar predicted metabolic profile within the collection of complete genomes in their database. In this case the genome is that of Clostridium novyi. Another organism used here for comparison was Methanobrevibacter smithii which co-occurs with C. minuta, has also been associated with lower BMI, but was not found to be highly heritable [2]. Since C. minuta was described, two other Christensenella species have been proposed and their genomes have been sequenced (C. timonensis and C. massiliensis) [17,18]. However, it is important to note that the descriptions of these two isolates were not done using accepted chemotaxonomic analyses and thus whether or not these should be considered true members of the Christensenella genus is unclear. Similarly, in the phylogenetic tree of all bacterial type strains (see below), Catabacter hongkongensis appears to group with the three sequenced Christensenella species, suggesting that further taxonomic revision in this group may be required.
In examination of the RAST annotation of the C. minuta DSM 22607 genome, the most striking characteristic to us was the large number of genes (n=409) predicted to be involved in carbohydrate utilization (Table 1). This is striking in that the total number is much higher than that seen for genomes of the close relatives we examined, with the exception of C. timonensis which has 434. In addition, the genome of the organisms flagged by RAST as having the most similar predicted metabolic profile, C. novyi, has much fewer (201) as does the genome of one of the key co-occurring organisms, M. smithii, which has only 90. There are many reasons why the two Christensenella species could have more carbohydrate metabolism genes annotated. One possibility is simply inaccuracies in annotation. But it could also be a reflection of true higher numbers of carbohydrate metabolizing genes in these species. We did not view these numbers of putative carbohydrate metabolism genes as being a precise measure of metabolism in these organisms but instead used this annotation as a means to focus our attention on these genes to see if any more light could be shed on possible unusual features of Christensenella.
C. minuta and selected organisms for comparison. Shown are the results from RAST annotation of these genomes.
| Isolate | Total Carbs | Monosaccharides | D-ribose | Accession |
|---|---|---|---|---|
| C. timonensis_Marseille_P2437 | 434 | 125 | 91 | GCF_900087015.1 |
| C. minuta_DSM_22607_UCD | 409 | 179 | 127 | GCA_001652705.1 |
| C. sp_AF73_05CM02 | 405 | 192 | 140 | GCF_001678845.1 |
| C. massiliensis_Marseille_P2438 | 300 | 110 | 77 | LT700187.1 |
| C. sp_Marseille_P3954 | 228 | 88 | 46 | GCF_900604345.1 |
| Caldicoprobacter oshimari | 218 | 74 | 7 | GCA_000526435.1 |
| Clostridium novyi | 201 | 41 | 7 | GCF_000014125.1 |
| Tindallia californiensis | 169 | 18 | 13 | GCA_900107405.1 |
| Methanobrevibacter smithii | 90 | 5 | 1 | GCF_000016525.1 |
RbaA/B/C genes
Examination of the predicted carbohydrate metabolism genes in more detail revealed a more striking finding. A significant fraction of all these genes in Christensenella (127 in total) were annotated as encoding homologs of RbsA, RbsB or RbsC which together make up the Ribose ABC Transport System. Early work focused on the importance of these genes in the transport of D-ribose [19,20], but there is more recent evidence that homologs of these genes may be involved in quorum sensing instead of ribose transport [21-24]. This is perhaps not surprising given the fact that the quorum sensing AI-2 autoinducer is derived from the ribosyl moiety of S-ribosylhomocysteine (a potential target of the transporters). Given the recently hypothesized role of AI-2 in structuring gut microbial communities [25] we hypothesize that the massive expansion of the Rbs transporters in this lineage may relate to quorum-sensing. Furthermore, this could relate to the demonstrated importance of this organism in structuring the gut microbial community. In particular, it could be that members of the Christensenella group act as “foundation” species in the human gut and that they directly structure the microbial community through quorum sensing mediated interactions.
In order to understand the expansion of these genes, we started by constructing a “whole genome tree” of all the available bacterial type strains at NCBI. We downloaded 9,499 type strain genomes and ran them through Phylosift in order to build a concatenated marker tree. Once the tree was constructed, we overlaid the number of RbsA, RbsB, and RbsC proteins onto the tips of the tree. A portion of this tree, containing the 372 strains phylogenetically closest to Christensenella (plus an outgroup, Streptococcus porcinus) is shown in Supplemental Figure 1. The portion of the tree containing the Christensenella clade is shown in Figure 1.
Phylogenetic relationships of Christensenella based on whole genome analysis. Shown is a zoom in of a larger phylogenetic tree of 9499 type strains generated from concatenated marker genes using Phylosift. Values at the node represent bootstrap values from RaxML.
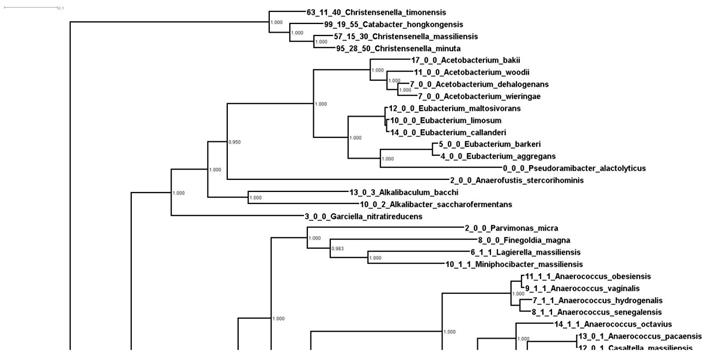
In Figure 1 we can see firstly that the Christensenella clade is not particularly phylogenetically close to the other bacteria in this tree. In fact the group is an outgroup to all other 368 strains in this tree (Supplemental Figure 1). Additionally, most of the bacteria in this tree encode relatively few RbsA/B/C homologs. There are multiple possible explanations for the pattern seen in number of these genes. These include lateral gene transfer into or duplication events in the ancestry of species with more of these genes, or alternatively, ancestral events such as these and then loss in lineages with fewer numbers. We consider it most likely that there was an expansion (either by duplication or lateral gene transfer) of these genes within the Christensenella clade (and a few other lineages).
As another way of looking at this data, we performed a BLAST-p [26] search (e-value cutoff of 1e -30) using all 127 RbsA/B/C sequences from C. minuta DSM22607 against all 9,499 downloaded type strain genomes. The 10 organisms with the most RbsA/B/C predicted proteins matching at an e-value of 1e-30 or better in their genomes from this search are listed in Table 2, along with C. timonensis and C. massiliensis (duplicate genomes from the same strains were removed). While there are not many homologs of any of the RbsA/B/C proteins present in the close relatives to C. minuta, there are a few other distantly related bacteria (Rhizobiales) with even more homologs. However, the possession of a large number of RbsB gene copies appears unique to the Christensenella lineage. When all 9,499 genomes are ranked by the number of RbsB homologs alone, the top 10 species are all Christensenella, Catabacter, and one Clostridium bolteae isolate (data not shown).
The 10 organisms with the most RbsA/B/C predicted proteins using our approach
| Rank | Genome | RbsA | RbsB | RbsC | Total | |
|---|---|---|---|---|---|---|
| 1 | Kaistia_algarum | 140 | 6 | 85 | 231 | GCF_002930635.1 |
| 2 | Mesorhizobium_helmanticense | 130 | 7 | 66 | 203 | GCA_003034915.1 |
| 3 | Mesorhizobium_sanjuanii | 125 | 7 | 62 | 194 | GCF_002529485.1 |
| 4 | Christensenella_sp._AF73-05CM02 | 104 | 27 | 55 | 186 | GCF_002529485.1 |
| 5 | Catabacter_hongkongensis_strain_HKU16 | 99 | 19 | 55 | 173 | GCA_000981035.1 |
| 6 | Christensenella_minuta_strain_DSM_22607 | 95 | 28 | 50 | 173 | GCA_001652705.1 |
| 7 | Rhizobium_album | 111 | 4 | 57 | 172 | GCF_003122325.1 |
| 8 | Bauldia_litoralis | 102 | 4 | 60 | 166 | GCF_900104485.1 |
| 9 | Mesorhizobium_ephedrae | 107 | 6 | 46 | 159 | GCF_003012745.1 |
| 10 | Kaistia_granuli | 100 | 4 | 54 | 158 | GCF_000380505.1 |
| 49 | Christensenella_timonensis | 63 | 11 | 40 | 114 | GCF_900087015.1 |
| 71 | Christensenella_massiliensis | 57 | 15 | 30 | 102 | GCA_900155415.1 |
Since we submitted the genome sequence of C. minuta DSM 22607 to NCBI in 2016, the same strain (DSMZ 22607) has had its genome sequence deposited several more times. In order to ensure that the results reported here were not due to sequencing or assembly artifacts we compared all these DSMZ 22607 assemblies themselves as well as the RAST annotations of the genes of interest. The average nucleotide identity (ANI) between assemblies of this strain was >99.98% demonstrating that they are virtually identical. We found the same with the RAST results, where the number of annotated Rbs transporters was between 126-128 per genome in every case.
Given the potential importance (positive and negative) of C. minuta in human health, and the fact that interventions based on this bacteria are already being considered, genomic analysis paired with functional experiments will be critical for a deeper understanding of this organism. In particular, the apparent expansion of genes encoding RbsA/B/C in this lineage warrants investigation.
Nucleotide sequence accession numbers
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession LWGY00000000. The version described in this paper is version LWGY01000000.
Supplementary Material
Supplemental Figure 1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Morotomi M, Nagai F, Watanabe Y. Description of Christensenella minuta gen. nov, sp. nov, isolated from human faeces, which forms a distinct branch in the order Clostridiales, and proposal of Christensenellaceae fam. nov. Int J Syst Evol Microbiol. 2012;62:144-149
2. Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R. et al. Human genetics shape the gut microbiome. Cell. 2014;159:789-799
3. Alonso BL, Irigoyen von Sierakowski A, Sáez Nieto JA, Rosel AB. First report of human infection by Christensenella minuta, a gram-negative, strickly anaerobic rod that inhabits the human intestine. Anaerobe. 2017;44:124-125
4. Waters JL, Ley RE. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019;17:83
5. Rosa BA, Hallsworth-Pepin K, Martin J, Wollam A, Mitreva M. Genome Sequence of Christensenella minuta DSM 22607T. Genome Announc. 2017:5 doi:10.1128/genomeA.01451-16
6. Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 2015;31:587-589
7. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75
8. Darling AE, Jospin G, Lowe E, Matsen FA 4th, Bik HM, Eisen JA. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2014;2:e243
9. Kiełbasa SM, Wan R, Sato K, Horton P, Frith MC. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21:487-493
10. Zhang Z, Wood WI. A profile hidden Markov model for signal peptides generated by HMMER. Bioinformatics. 2003;19:307-308
11. Price MN, Dehal PS, Arkin AP. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490
12. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312-1313
13. Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119
14. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403-410
15. Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114
16. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043-1055
17. Ndongo S, Dubourg G, Khelaifia S, Fournier P-E, Raoult D. Christensenella timonensis, a new bacterial species isolated from the human gut. New Microbes New Infect. 2016;13:32-33
18. Ndongo S, Khelaifia S, Fournier P-E, Raoult D. Christensenella massiliensis, a new bacterial species isolated from the human gut. New Microbes New Infect. 2016;12:69-70
19. Lopilato JE, Garwin JL, Emr SD, Silhavy TJ, Beckwith JR. D-ribose metabolism in Escherichia coli K-12: genetics, regulation, and transport. J Bacteriol. 1984;158:665-673
20. Iida A, Harayama S, Iino T, Hazelbauer GL. Molecular cloning and characterization of genes required for ribose transport and utilization in Escherichia coli K-12. J Bacteriol. 1984;158:674-682
21. Taga ME, Semmelhack JL, Bassler BL. The LuxS-dependent autoinducer AI-2 controls the expression of an ABC transporter that functions in AI-2 uptake in Salmonella typhimurium: Autoinducer-dependent gene regulation. Mol Microbiol. 2008;42:777-793
22. James D, Shao H, Lamont RJ, Demuth DR. The Actinobacillus actinomycetemcomitans ribose binding protein RbsB interacts with cognate and heterologous autoinducer 2 signals. Infect Immun. 2006;74:4021-4029
23. Shao H, James D, Lamont RJ, Demuth DR. Differential interaction of Aggregatibacter (Actinobacillus) actinomycetemcomitans LsrB and RbsB proteins with autoinducer 2. J Bacteriol. 2007;189:5559-5565
24. Armbruster CE, Pang B, Murrah K, Juneau RA, Perez AC, Weimer KED. et al. RbsB (NTHI_0632) mediates quorum signal uptake in nontypeable Haemophilus influenzae strain 86-028NP. Mol Microbiol. 2011;82:836-850
25. Thompson JA, Oliveira RA, Djukovic A, Ubeda C, Xavier KB. Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep. 2015;10:1861-1871
26. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K. et al. BLAST: architecture and applications. BMC Bioinformatics. 2009 p. 421. doi:10.1186/1471-2105-10-421
Author contact
![]() Corresponding author: Email: Jonathan Eisen, jaeisenedu.
Corresponding author: Email: Jonathan Eisen, jaeisenedu.