ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2015; 3:36-39. doi:10.7150/jgen.9204 This volume Cite
Short Research Communication
Evaluation of the Performances of Ribosomal Database Project (RDP) Classifier for Taxonomic Assignment of 16S rRNA Metabarcoding Sequences Generated from Illumina-Solexa NGS
Giovanni Bacci1,2, Alessia Bani1, Marco Bazzicalupo1, Maria Teresa Ceccherini3, Marco Galardini1*, Paolo Nannipieri3, Giacomo Pietramellara3, Alessio Mengoni1 ![]()
1. Department of Biology, University of Florence, via Madonna del Piano 6, I-50019 Sesto Fiorentino, Firenze, Italy.
2. Consiglio per la Ricerca e la Sperimentazione in Agricoltura, Centro di Ricerca per lo Studio delle Relazioni tra Pianta e Suolo (CRA-RPS), Via della Navicella 2/4, I-00184 Roma, Italy.
3. Department of Agrifood Production and Environmental Science, University of Florence, P.le delle Cascine 28, I-50144, Firenze, Italy.
* Present address: EMBL-EBI - European Bioinformatics Institute Wellcome Trust Genome Campus Hinxton, Cambridge CB10 1SD, UK.
Published 2015-2-1
Abstract
Here we report a benchmark of the effect of bootstrap cut-off values of the RDP Classifier tool in terms of data retention along the different taxonomic ranks by using Illumina reads. Results provide guidelines for planning sequencing depths and selection of bootstrap cut-off in taxonomic assignments.
Keywords: 16S rRNA, metabarcoding, ribosomal database project, OTU clustering, bacterial communities.
Introduction
The use of 16S rRNA massive sequencing has deeply improved the technical possibilities to describe the taxonomic composition and functionality of microbial communities [1]. Following the reduction in DNA sequencing cost, many studies have been performed using amplicon libraries to taxonomically describe microbial communities in many different environments. The large number of sequence reads can be taxonomically assigned by comparison with taxonomically classified sequences present in dedicated databases of 16S rRNA genes, as for instance SILVA [2], Greengenes [3] or the Ribosomal Database Project [4]. In particular, one of the most popular tools used to assign sequence reads to the prokaryotic taxonomy is the Naïve Bayesian Classifier tool hosted by Ribosomal Database Project (RDP Classifier) [4]. The RDP Classifier tool uses a very fast algorithm, based on the Bayes' theorem, suitable for the analysis of large amount of sequence data. This algorithm has been tested on near-full-length 16S rRNA sequences and on randomly generated 16S rRNA sequence fragments of 400, 200, 100 and 50 bases in length from a number (5,014) of type strains belonging to 988 genera [4]. An overall accuracy of above 88.7% and 83.2% for 400 and 200 base segments, respectively (very similar to the accuracy obtained with the near-full-length 16S rRNA sequence) was reported [4]. Moreover, an average accuracy at genus level of 71.1% and 51.5% for the 100 and the 50 base segments respectively was found. However, these results have been obtained using a 16S rRNA sequence fragments dataset built with sequences derived from well taxonomically defined organisms. No data have been reported on datasets composed by 16SrRNA sequences from particular regions of the 16S molecule (e.g. V3 and V6) and obtained after amplification of DNA from environmental samples.
In the last years, Illumina sequencing technology has emerged as one of the most popular sequencing technology, thanks to the lower prices, higher number of generated sequences and accuracy than pyrosequencing and Ion Torrent technologies [1, 5-8]. In particular, different Illumina platforms are available (with different cost of sequencing) which provide different number of reads and different reads lengths (see for instance http://www.illumina.com/systems/sequencing.ilmn). Additionally, Illumina reads are usually 100-200 nt long (depending on the techniques used) and 16S rRNA amplicon studies have focused on single variable regions of the 16S rRNA gene, as the V3, V4 or V6, which are approximately 100-300 bp long. Consequently, a concern about the amount of reads to generate and the setting of the bootstrap threshold of RDP Classifier to provide biologically meaningful data is present. More specifically, there is a lack of information on the percentage of reads which can be assigned to the various phylogenetic levels with Illumina 16S rRNA metabarcoding.
Here, we report a benchmark of RDP Classifier based on environmental sequence datasets obtained with Illumina sequencing technology. In particular, we investigated the effect of bootstrap cutoff values on the accuracy of taxonomic attribution of Illumina reads. Results obtained provide a guideline for the selection of optimal bootstrap cutoff values in terms of data retention along the different taxonomic ranks.
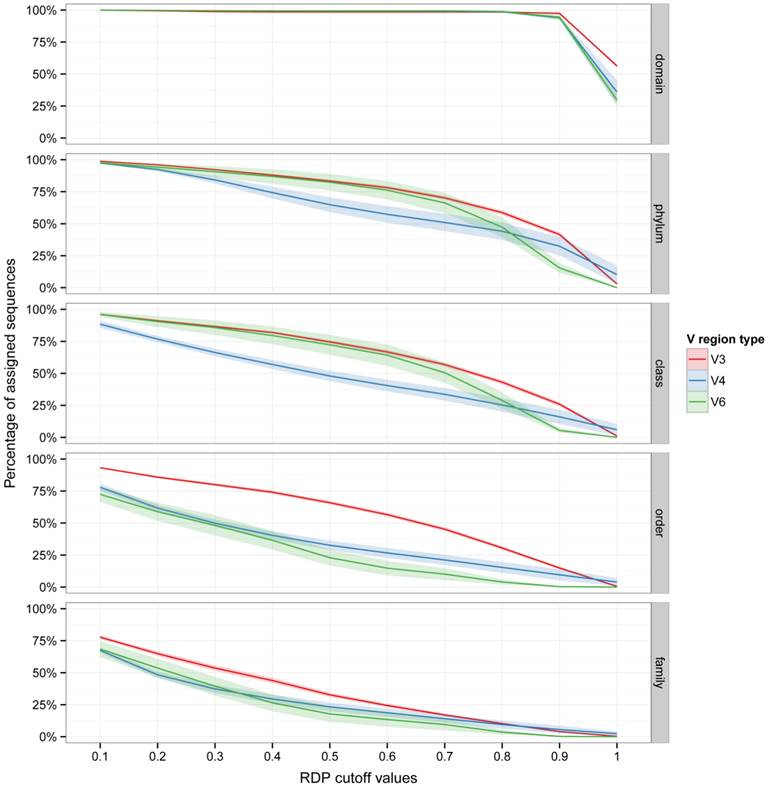
Five datasets of 16S rRNA gene Illumina reads, generated from environmental DNA, were analyzed (Table 1). These datasets contains a high number of reads per sample (from 28634 to 759518 reads per sample) and are including reads obtained from V3, V4 and V6 regions. Reads present in the analyzed datasets were trimmed with StreamingTrim version 1.0 [9], before taxonomic assignment with the RDP Classifier. The proportion of assigned reads in relation to the bootstrap cutoff value (from 0.1 to 1.0 with an increment of 0.1) for each taxonomic level (from domain to genus) is reported in Figure 1. As expected, the proportion of assigned reads decreased going down along taxonomic levels from phylum (from a mean of 100% to a mean of 25%, in the two datasets) to genus (from a mean of 60% to a mean of smaller than 5%, in the two datasets). In particular it is worth noticing that all datasets, which included three variable regions (V3, V4 and V6) of 16S rRNA gene, more than 25% of the reads could be assigned to the family level using a bootstrap cutoff value of 0.5 (the default cut-off value reported in the RDP Classifier tutorial). Moreover, even at higher cutoff values (> 0.8) an appreciable number of reads were still assigned (5%-10%). Interestingly, the V3 region performed better in the taxonomic attribution at Order and Family levels, indicating that even highly stringent bootstrap cut-off values (e.g. 0.7) may allow to assign more reads from V3 region than from V4 and V6 region, which consequently resulted less taxonomically informative.
Description of the datasets used*.
| BioProject* | rRNA region | Average reads length | Number of samples | Average number of sequences per sample | Environment |
|---|---|---|---|---|---|
| PRJEB6047 | V3 | 302bp | 72 | 61023 | Subgingival, supragingival, and tongue plaque from healthy and periodontal subjects |
| PRJNA245381 | V3 | 300bp | 100 | 28634 | Soil contaminated with increasing level of ionic Ag |
| PRJNA217938 | V4 | 288bp | 25 | 476230 | Samples from the surface to depth in Upper Mystic Lake, Winchester, MA |
| PRJNA238275 | V4 | 251bp | 6 | 759518 | Soil associated with the rhizosphere of the coffee plant (Coffea canephora) in Brazil |
| PRJNA188383 | V6 | 200bp | 48 | 66887 | Seawater and surface sediments retrieved from the Arctic Ocean |
* The ID of the accession (http://www.ncbi.nlm.nih.gov/bioproject/), the variable region sequenced, the type of reads, the number of different samples analyzed and the number of reads is shown.
Effect of bootstrap cut-off thresholds on the number of reads. The percentage of trimmed reads assigned to each taxonomic level is reported versus RDP bootstrap cut-off values. Shaded lines correspond to the 95% confidence interval assuming normality.
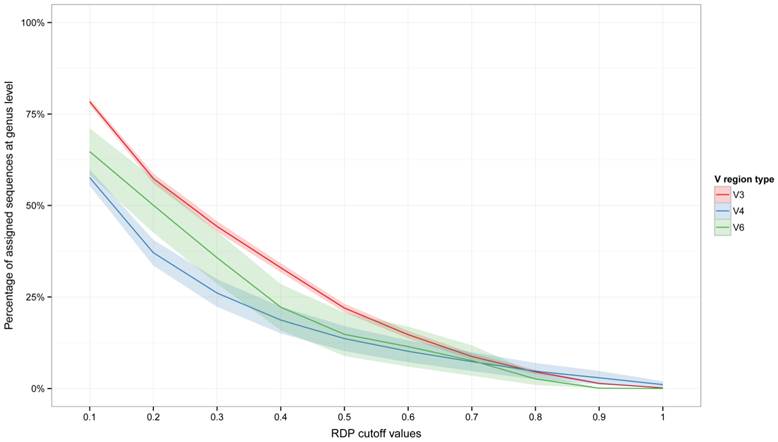
The assignments trend at the genus level (the lower taxonomic level that can be obtained using the RDP Classifier) was then inspected (Figure 2). Here also, V3 region better performed than V4 and V6 region in the retention of taxonomic information lowering bootstrap values, especially at bootstrap cutoff value of 0.5 and lower. A pseudo-fit of curve was also produced (Supplementary Figure 1), which may allow researchers to infer the percentage of sequences that could be assigned to the genus level at different RDP bootstrap cut-offs.
Percentage of assigned reads with respect to bootstrap cut-off thresholds at the genus level. Plots report the assigned reads for all dataset analyzed. Shaded lines correspond to the 95% confidence interval assuming normality.
In conclusion, Illumina reads, shorter than 200 nt, can be classified using one of the most common 16S rRNA sequence classifier: the RDP Classifier. As one would expect, the increase of the bootstrap cutoff value leads to a decreased number of assigned sequences. However, even at cutoffs higher than those indicated in the RDP Classifier tutorial, approximately 20-30% of the analyzed reads were still assigned. These results indicate that Illumina-based metabarcode sequencing of 16S rRNA gene can provide reliable information for taxonomic composition of a community at the genus level even using classification software not specifically designed for this type of sequences. The reported models for trend plots can guide experimentalists in choosing the sequencing depth more adapted for retaining an appreciable number of assigned reads different taxonomic resolution.
Supplementary Material
Supplementary Figure 1Acknowledgements
This work was supported by a grant from the Ente Cassa di Risparmio di Firenze (Grant n° 2010/4384 “Centro di Metagenomica del suolo”) and by a fellowship to GB from MIPAAF.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Degnan PH, Ochman H. Illumina-based analysis of microbial community diversity. Isme Journal. 2012;6(1):183-194
2. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research. 2013;41(D1):D590-D596
3. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Applied and Environmental Microbiology. 2006;72(7):5069-5072
4. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Applied and Environmental Microbiology. 2007;73(16):5261-5267
5. Gloor GB, Hummelen R, Macklaim JM, Dickson RJ, Fernandes AD, MacPhee R, Reid G. Microbiome Profiling by Illumina Sequencing of Combinatorial Sequence-Tagged PCR Products. PLoS ONE. 2010;5(10):e15406
6. Bartram AK, Lynch MDJ, Stearns JC, Moreno-Hagelsieb G, Neufeld JD. Generation of Multimillion-Sequence 16S rRNA Gene Libraries from Complex Microbial Communities by Assembling Paired-End Illumina Reads. Applied and Environmental Microbiology. 2011;77(11):3846-3852
7. Claesson MJ, Wang Q, O'Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O'Toole PW. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Research. 2010 38(22)
8. Salipante SJ, Kawashima T, Rosenthal C, Hoogestraat DR, Cummings LA, Sengupta DJ, Harkins TT, Cookson BT, Hoffman NG. Performance Comparison of Illumina and Ion Torrent Next-Generation Sequencing Platforms for 16S rRNA-Based Bacterial Community Profiling. Applied and Environmental Microbiology. 2014;80(24):7583-7591
9. Bacci G, Bazzicalupo M, Benedetti A, Mengoni A. StreamingTrim 1.0: a Java software for dynamic trimming of 16SrRNA sequence data from metagenetic studies. Molecular Ecology Resources. 2014Mar;14(2):426-34 doi: 10.1111/1755-0998.12187
10. Masella A, Bartram A, Truszkowski J, Brown D, Neufeld J. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13(1):31
Author contact
![]() Corresponding author: Alessio Mengoni, e-mail: alessio.mengoniit.
Corresponding author: Alessio Mengoni, e-mail: alessio.mengoniit.