ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2023; 11:40-44. doi:10.7150/jgen.86558 This volume Cite
Short Research Paper
Genomic analysis of Chlamydia psittaci from a multistate zoonotic outbreak in two chicken processing plants
Bernard J. Wolff1, Jessica L. Waller1, Alvaro J. Benitez1, Anna Gaines2, Andrew B. Conley2, Lavanya Rishishwar2,3, Aroon T. Chande2,3, Shatavia S. Morrison1, I. King Jordan2,3, Maureen H. Diaz1, Jonas M. Winchell1, ![]()
1. Division of Bacterial Diseases, Centers for Disease Control and Prevention, Atlanta, GA, USA
2. Applied Bioinformatics Laboratory, Atlanta, GA, USA
3. School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, USA
Received 2023-5-27; Accepted 2023-7-27; Published 2023-8-19
Abstract

Four Chlamydia psittaci isolates were recovered from clinical specimens from ill workers during a multistate outbreak at two chicken processing plants. Whole genome sequencing analyses revealed high similarity to C. psittaci genotype D. The isolates differed from each other by only two single nucleotide polymorphisms, indicating a common source.
Chlamydia psittaci is an obligate intracellular bacterium with a unique biphasic life cycle that alternates between an infectious non-replicating elementary body (EB) form and a metabolically active replicating reticulate body (RB) form [1]. This species is a member of the Chlamydiace family, which dates back to 50 to 250 million years ago, and has been shown to have a broad range of host species [2]. While C. psittaci has traditionally been documented in psittacine birds, it has also been found in cattle, sheep, swine, goats, cats, horses, and at least 465 bird species spanning 30 different orders [2-4]. It is the etiologic agent of psittacosis, a respiratory zoonotic infection in humans that can vary in severity and have multi-organ involvement, resulting in significant morbidity and mortality [5]. While C. psittaci has been documented in turkeys for decades, evidence of its presence in chickens has only been recently documented [6, 7]. There are ten well described C. psittaci genotypes designated A-G, E/B, M56 and W/C, which differ in virulence potential and host species tropism [2, 8]. Genotype D is most commonly associated with poultry outbreaks and is extensively excreted relative to other genotypes [2, 6, 8, 9]. While there are many publicly available genome sequences of C. psittaci, there are no whole genome sequences available for C. psittaci isolates originating from chickens. We performed whole genome sequencing analysis on isolates obtained from clinical specimens collected from ill workers during an outbreak at two chicken processing plants in 2018.
From August 31 to September 28, 2018, 50 chicken processing plant workers in Virginia and 30 in Georgia became ill with a respiratory illness, 29 of whom required hospitalization [10]. Sputum, bronchoalveolar lavage washings (BAL), nasopharyngeal (NP) and oropharyngeal (OP) swabs, and stool specimens from plant workers meeting the case definition were submitted to the Centers for Disease Control and Prevention (CDC) and tested by real-time polymerase chain reaction (PCR) [10, 11]. Thirteen patients had C. psittaci detected in one or more clinical specimens [11]. Sequencing of the outer membrane protein A (ompA) gene was attempted on specimens with Ct values less than 30 (n=7) as previously described [11, 12]. All specimens matched the ompA gene of genotype D with 100% pairwise identity. Culture was attempted on lower respiratory tract specimens (n=8) that were PCR positive, owing to the relatively higher C. psittaci concentrations previously found in this specimen type after inoculation of Vero cell monolayers [13]. Isolates were obtained from four of eight specimens, with recovery rate being affected by bacterial and/or fungal contamination, lower pathogen load, and possible low viability due to transport and cell culture delays. Growth of C. psittaci was confirmed by observing cytopathogenic effects through microscopy and concomitant real-time PCR testing of cell culture supernatants [11]. Chlamydia EBs were purified by renografin gradient centrifugation as previously described [14]. Nucleic acid was extracted from the purified EBs using the MasterPure Complete DNA and RNA Purification Kit (Epicentre, Madison, WI) according to the manufacturer's instructions. Libraries were prepared using the Illumina Nextera XT library preparation kit per manufacturer's instructions and sequenced on a MiSeq System (Illumina, San Diego, CA) with version 2 500 cycle kits using 2x250 paired-end sequencing. Long read sequencing was performed using either Pacific Biosciences (PacBio) RS II (Menlo Park, CA) or Oxford Nanopore MinION (OX4 4DQ, UK) using Oxford Nanopore Rapid PCR Barcoding Kit (SQK-RPB004). Libraries were prepared using the PacBio SMRTbell Template Prep Kit 1.0 or the Oxford Nanopore Rapid PCR Sequencing Kit per the manufacturer's instructions. Long read sequencing was not performed for one isolate, VA1, due to contamination; however, Illumina MiSeq sequence data yielded a sufficiently high-quality genome for this isolate. PacBio reads for GA1, VA2 and VA3 were assembled using HGAP version 3, Oxford Nanopore MinION reads for GA1 were assembled using Flye [15, 16], Medaka [17], and Pilon [18], and Illumina reads for VA1 were assembled using SPAdes version 3.11.1 [19]. PacBio and MinION assemblies yielded finished chromosomal assemblies, while assembly of Illumina reads resulted in a nearly complete genome with 3 contigs (>1,000bp). No major differences were observed in the finished PacBio and Nanopore assemblies for the GA1 isolate. For whole genome comparisons, we used the PacBio assemblies for GA1, VA2, and VA3 and the draft Illumina assembly for VA1 (Table 1).
Sequencing metadata for the four outbreak isolates.
| Isolate Name | GA1 | VA1 | VA2 | VA3 |
|---|---|---|---|---|
| Accession Number | JARFVI000000000 | JARFVH000000000 | JARFVG000000000 | JARFVF000000000 |
| Location | Georgia | Virginia | Virginia | Virginia |
| Illumina MiSeq Sequencing | ✔ | ✔ | ✔ | ✔ |
| ONT Sequencing | ✔ | X | X | X |
| PacBio RSII Sequencing | ✔ | X | ✔ | ✔ |
| Percent of Genome Covereda | 100 | 100 | 100 | 100 |
| # Contigsb | 1 | 3 | 1 | 1 |
| Genome Sizeb | 1161591 | 1163341 | 1161755 | 1161426 |
| N50b | 1161591 | 775497 | 1161755 | 1161426 |
aPercent of reference genome NJ1 covered by Illumina reads
bFinal assemblies used for whole genome comparison
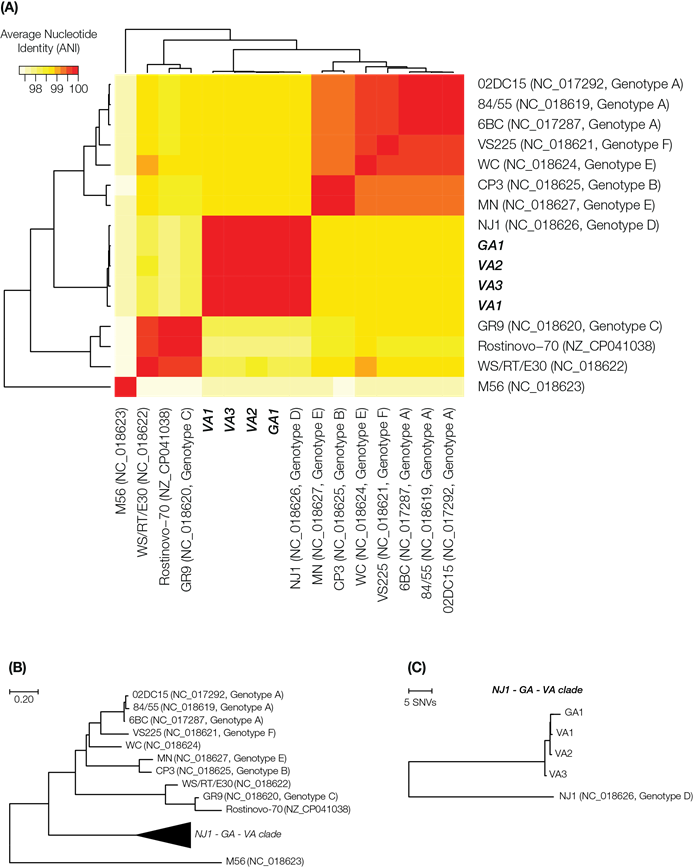
Each of the assembled genomes was compared to reference C. psittaci assemblies from NCBI using all-against-all pairwise average nucleotide identity (ANI) values, calculated by using the MUMmer program [20]. All the isolates form a cluster with the reference genome NJ1, a C. psittaci genotype D isolate (NC_018626.1; Figure 1A). All the isolates were observed to be >99% similar to NJ1. ANI values were converted to distance measures (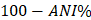 ) and used to construct a phylogenetic tree using the Neighbor-Joining algorithm [21] in the program Mega X [22]. The phylogenetic comparison also supported clustering of the isolates with the NJ1 reference genome (Figure 1B). NJ1 was subsequently used throughout the study as the reference genome when performing additional analyses on these newly assembled genomes.
) and used to construct a phylogenetic tree using the Neighbor-Joining algorithm [21] in the program Mega X [22]. The phylogenetic comparison also supported clustering of the isolates with the NJ1 reference genome (Figure 1B). NJ1 was subsequently used throughout the study as the reference genome when performing additional analyses on these newly assembled genomes.
Comparison of whole genome sequences of outbreak-associated C. psittaci isolates and reference genomes. (A) Whole genome average nucleotide distance phylogenetic tree. (B) Average nucleotide identity heatmap with all publicly available reference genomes of C. psittaci and the four outbreak isolates.
Whole genome alignment using Mauve version 20150226 build 10 [23] demonstrated no major rearrangements between the assemblies and NJ1 (Supplementary Figure 1). Comparison of finished assemblies to the reference genome indicated a 361 bp insertion within the membrane protein gene (WP_014946495.1). This insertion was also detected in the genome of an additional reference isolate WC, indicating a deletion in NJ1 not shared by the isolates analyzed in this report. For variant analysis, Illumina MiSeq reads were mapped against the C. psittaci NJ1 genome using the program BWA [24] followed by variant calling with SAMtools [25, 26]. The four outbreak isolates are nearly identical to each other, differing by at most two SNPs and are also highly similar to the NJ1 reference with only 61 SNPs and 2 insertions identified (Table 2).
Number of SNPs per genome identified by pairwise comparison of outbreak isolates and reference NJ1.
| NJ1 | GA1 | VA1 | VA2 | VA3 | |
|---|---|---|---|---|---|
| NJ1 | 63 | 62 | 61 | 62 | |
| GA1 | 63 | 1 | 2 | 1 | |
| VA1 | 62 | 1 | 2 | 1 | |
| VA2 | 61 | 2 | 2 | 2 | |
| VA3 | 62 | 1 | 1 | 2 |
Supplementary Table 1 lists all the observed nucleotide substitutions, positions, variant qualities, affected genes, and variant effects in these isolates. Variants were categorized by potential impact based on the nature of the mutation [27]. One insertion and one variant were considered high impact because they resulted in a frameshift mutation and the loss of a stop codon. However, these mutations occurred in a hypothetical protein and a domain of unknown function, so the potential disruption of these mutations remains unclear. Thirty variants were classified as moderate variants causing missense mutations within genes. There were ten variants and one insertion that were intergenic modifying variants resulting in upstream gene variants in regulatory regions upstream of genes. Lastly, twenty variants resulted in a synonymous variant and thus were classified as low impact.
The isolates in this study were remarkably conserved relative to the genotype D reference strain NJ1, which was initially isolated from a turkey in 1954 [2]. Despite nearly 65 years between the original isolation and multiple passages of the original reference strain, the genomes of the outbreak isolates gained the equivalent of one variant per year since the original isolation of the reference genotype strain. However, due to the lack of representative genomes it cannot be determined how or when these variants were accumulated. In contrast to other intracellular bacterial species that demonstrate ongoing genome reduction and frequent horizontal gene transfer [19], our findings are consistent with the reported stability of Chlamydial genomes over time despite the ability to frequently jump host species [2]. C. psittaci has been well documented in turkeys with genotype D being the most prevalent. Chickens have only recently been studied for the presence of C. psittaci in Europe, Australia and Asia [3, 6, 28-31], but to date no studies of its prevalence in chickens in the United States have been published. It is possible one or more of the variants identified in the outbreak isolates allowed genotype D to more easily infect chickens. This phenomena has been documented previously in other bacterial species; Viana et al. determined that one variant within the dltB gene responsible for lipoteichoic acid biosynthesis, an upstream precursor to cell wall biosynthesis, was solely responsible for the Staphylococcus aureus human-to-rabbit host jump over 40 years ago [32]. Mutations impacting membrane proteins are of particular interest due to their interaction with host cell receptors, but other genes may also contribute to host specificity. Of the variants identified in this study that were of moderate impact or modifiers in intergenic DNA, two impact a perforin family protein and one is located in an outer membrane protein. All three variants could potentially impact host preference. Further research will be needed to investigate the infectivity of these isolates in chickens relative to other genotype D isolates.
Traditionally, it was conjectured that chickens are less susceptible to C. psittaci compared to turkeys, although recent outbreaks have brought this into question [6, 7]. Differences in turkey and chicken processing operations were thought to be a contributing reason behind the lower observed rates of C. psittaci infection in chickens. Turkeys are typically raised for 15 to 17 weeks before they are slaughtered compared to 6 weeks for a typical chicken [3]. The extended time in turkeys could allow C. psittaci to spread to a larger portion of the population and have more time to reach higher bacterial loads in the birds before slaughter. However, up until 2010 the only C. psittaci genotypes detected in chickens were the less virulent genotypes B, C, and E/B [7, 29, 33]. Genotype D has been shown to be highly virulent and excreted extensively compared to other genotypes and has only recently been documented in chickens [3, 6]. These characteristics may allow genotype D to more rapidly infect flocks in the close quarters of chicken houses. This genotype was identified early in the outbreak using sanger sequencing of the ompA gene following nucleic acid extraction from clinical specimens. From the initial ompA sequencing and knowing both plants were receiving chickens from the same farms, we were able to hypothesize that the outbreaks in both Georgia and Virginia were likely from the same source, and that it was a historically highly virulent genotype that required aggressive treatment and management to mitigate the outbreak. Additional ompA sequencing with next generation and third generation sequencing chemistries provided additional clues and insights into how C. psittaci may be evolving to more efficiently infect hosts and/or increase virulence and should be considered when investigating uncommon outbreaks.
Supplementary Material
Supplementary figure and table.
Competing Interests
The authors have declared that no competing interest exists.
References
1. AbdelRahman YM, Belland RJ. The Chlamydial Developmental Cycle. FEMS Microbiology Reviews. 2005;29:949-59
2. Read TD, Joseph SJ, Didelot X, Liang B, Patel L, Dean D. Comparative Analysis of Chlamydia psittaci Genomes Reveals the Recent Emergence of a Pathogenic Lineage with a Broad Host Range. MBio. 2013;4:e00604-12
3. Dickx V, Geens T, Deschuyffeleer T, Tyberghien L, Harkinezhad T, Beeckman DSA. et al. Chlamydophila psittaci Zoonotic Risk Assessment in a Chicken and Turkey Slaughterhouse. Journal of clinical microbiology. 2010;48:3244-50
4. Hotzel H, Berndt A, Melzer F, Sachse K. Occurrence of Chlamydiaceae spp. in a wild boar (Sus scrofa L.) population in Thuringia (Germany). Veterinary Microbiology. 2004;103:121-6
5. Balsamo G, Maxted AM, Midla JW, Murphy JM, Wohrle R, Edling TM. et al. Compendium of Measures to Control Chlamydia psittaci Infection Among Humans (Psittacosis) and Pet Birds (Avian Chlamydiosis), 2017. Journal of Avian Medicine and Surgery. 2017;31:262-82
6. Lagae S, Kalmar I, Laroucau K, Vorimore F, Vanrompay DJJomm. Emerging Chlamydia psittaci infections in chickens and examination of transmission to humans. Journal of medical microbiology. 2014;63:399-407
7. Gaede W, Reckling K-F, Dresenkamp B, Kenklies S, Schubert E, Noack U. et al. Chlamydophila psittaci Infections in Humans during an Outbreak of Psittacosis from Poultry in Germany. Zoonoses and Public Health. 2008;55:184-8
8. Van Lent S, Piet JR, Beeckman D, van der Ende A, Van Nieuwerburgh F, Bavoil P. et al. Full Genome Sequences of All Nine Chlamydia psittaci Genotype Reference Strains. Journal of Bacteriology. 2012;194:6930-1
9. Wolff BJ, Morrison SS, Pesti D, Ganakammal SR, Srinivasamoorthy G, Changayil S. et al. Chlamydia psittaci comparative genomics reveals intraspecies variations in the putative outer membrane and type III secretion system genes. Microbiology. 2015;161:1378-91
10. Shaw KA, Szablewski CM, Kellner S, Kornegay L, Bair P, Brennan S. et al. Psittacosis Outbreak among Workers at Chicken Slaughter Plants, Virginia and Georgia, USA, 2018. Emerg Infect Dis. 2019;25:2143-5
11. Wolff BJ, Morrison SS, Winchell JM. Development of a multiplex TaqMan real-time PCR assay for the detection of Chlamydia psittaci and Chlamydia pneumoniae in human clinical specimens. Diagnostic Microbiology and Infectious Disease. 2018;90:167-70
12. Mitchell SL, Wolff BJ, Thacker WL, Ciembor PG, Gregory CR, Everett KDE. et al. Genotyping of Chlamydophila psittaci by Real-Time PCR and High-Resolution Melt Analysis. 2009; 47: 175-81.
13. Schiller I, Schifferli A, Gysling P, Pospischil A. Growth characteristics of porcine chlamydial strains in different cell culture systems and comparison with ovine and avian chlamydial strains. The Veterinary Journal. 2004;168:74-80
14. Mukhopadhyay S, Clark AP, Sullivan ED, Miller RD, Summersgill JT. Detailed Protocol for Purification of Chlamydia pneumoniae Elementary Bodies. Journal of Clinical Microbiology. 2004;42:3288-90
15. Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nature Biotechnology. 2019;37:540-6
16. Lin Y, Yuan J, Kolmogorov M, Shen MW, Chaisson M, Pevzner PA. Assembly of long error-prone reads using de Bruijn graphs. Proceedings of the National Academy of Sciences. 2016;113:E8396-E405
17. Ltd ONT. medaka: Sequence correction provided by ONT Research
18. Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S. et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLOS ONE. 2014;9:e112963
19. Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A. et al. Assembling Single-Cell Genomes and Mini-Metagenomes From Chimeric MDA Products. Journal of Computational Biology. 2013;20:714-37
20. Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL. Alignment of whole genomes. Nucleic acids research. 1999;27:2369-76
21. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular biology and evolution. 1987;4:406-25
22. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Molecular biology and evolution. 2018;35:1547-9
23. Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Research. 2004;14:1394-403
24. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2009;25:1754-60
25. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics (Oxford, England). 2011;27:2987-93
26. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England). 2009;25:2078-9
27. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80-92
28. JUNJING YANG QY, JIANMING YANG, CHENG HE. Prevalence of avian Chlamydophila psittaci in China. Bulletin of the Veterinary Institute in Pulawy. 2007;51:347
29. Zhang F, Li S, Yang J, Pang W, Yang L, He C. Isolation and Characterization of Chlamydophila psittaci Isolated from Laying Hens with Cystic Oviducts. Avian Diseases. 2008;52:74-8
30. Laroucau K, Vorimore F, Aaziz R, Berndt A, Schubert E, Sachse K. Isolation of a new chlamydial agent from infected domestic poultry coincided with cases of atypical pneumonia among slaughterhouse workers in France. Infection, Genetics and Evolution. 2009;9:1240-7
31. Robertson T, Bibby S, O'Rourke D, Belfiore T, Agnew-Crumpton R, Noormohammadi AH. Identification of chlamydial species in crocodiles and chickens by PCR-HRM curve analysis. Veterinary Microbiology. 2010;145:373-9
32. Viana D, Comos M, McAdam PR, Ward MJ, Selva L, Guinane CM. et al. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nature Genetics. 2015;47:361-6
33. Vanrompay D, Butaye P, Sayada C, Ducatelle R, Haesebrouck F. Characterization of avian Chlamydia psittaci strains using omp1 restriction mapping and serovar-specific monoclonal antibodies. Research in Microbiology. 1997;148:327-33
Author contact
![]() Corresponding author: 1600 Clifton Rd. NE, MS G-03, Atlanta, GA 30333, United States. Telephone: (404) 639-4921; Fax: (404)718-1855. Email: jwinchellgov
Corresponding author: 1600 Clifton Rd. NE, MS G-03, Atlanta, GA 30333, United States. Telephone: (404) 639-4921; Fax: (404)718-1855. Email: jwinchellgov