ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2022; 10:33-38. doi:10.7150/jgen.68220 This volume Cite
Research Paper
Identification of the c.829_832delAATA Deletion Variants in the BRCA1 Gene Associated with Hereditary Breast/Ovarian Cancer ˗ Case Report
Malgorzata Ostrowska1 ![]() , Karolina Olszewska-Bozek2, Justyna Podlodowska3, Jadwiga Sierocinska-Sawa4, Jacek Wojcierowski2
, Karolina Olszewska-Bozek2, Justyna Podlodowska3, Jadwiga Sierocinska-Sawa4, Jacek Wojcierowski2
1. Department of Biotechnology, Microbiology and Human Nutrition, University of Life Sciences in Lublin, 8 Skromna St., 20-704 Lublin, Poland
2. Laboratory of Genetic Investigations, B. Dobrzanskiego 7 St., 20-262 Lublin
3. Center of Oncology of the Lublin Region St. Jana z Dukli, 7 K. Jaczewskiego St., 20-090 Lublin
4. Pathomorphology Department of Clinical Hospital I, I Independent Clinical Hospital, Stanislawa Staszica 16 St., 20-081 Lublin
Received 2021-10-19; Accepted 2022-1-26; Published 2022-2-14
Abstract

Determination of the BRCA1/BRCA2 mutation status in patients with breast and/or ovarian cancer is commonly performed using various molecular techniques. The use of targeted PCR-based tests only may not be sufficient, as not all possible variants are investigated. In the present study, we used next-generation sequencing (NGS) techniques to identify novel pathogenic variants in BRCA1 and BRCA2.
In this study, material (blood and FFPE) collected from a 67-year-old patient with ovarian cancer was used. The presence of hereditary mutations characteristic for the Polish population was examined using Sanger sequencing. BRCA1 and BRCA2 gene exons were amplified using the Devyser BRCA kit and sequenced on the Miniseq. No germline mutations characteristic for the Polish population were detected. However, 12 single nucleotide variants and 2 indels were identified. We found a new deleterious mutation of gene BRCA1 (c.829_832delAATA). To our knowledge, this mutation has not been reported yet in the Polish population and elsewhere. The use of the NGS technique increases the possibility of detecting mutational changes in patients with ovarian and/or breast cancer. Quick determination of pathogenic variants is important to facilitate specific therapy, in addition to the identification of familial predisposition to cancer.
Keywords: BRCA1, BRCA2, ovarian/breast cancer, NGS, PARP inhibitors
Introduction
Ovarian cancer was the eighth most common neoplasm in women and the 18th of all malignancies in the world in 2018 [1]. In 2018, there were nearly 295,400 new cases of ovarian cancer in the world and 5,077 in Poland [2]. Approximately, 15% of ovarian cancers are caused by germline BRCA1/2 (Breast cancer type 1 susceptibility protein and Breast cancer type 2 susceptibility protein) genes mutations [3], while 3% to 9% are related to pathogenic somatic variants [4].
The BRCA1 gene (OMIM#13705) is located on chromosome 17q12. It encodes a multifunctional protein involved in homologous recombination DNA repair [5], G2 cell cycle checkpoint regulation [6], cell survival, and chromosome stability [7]. The RING finger domain binds Bard1.p and Ola1.p forming an E3 ubiquitin ligase complex [8].
Germline mutations of the BRCA1 gene have been detected in the majority of familial breast and ovarian cancers. Additionally, 30% to 40% of sporadic cases are associated with altered expression of BRCA1 [9]. In the Polish population, the most commonly studied mutations in the BRCA1 gene include: 5382insC (c.5266dupC; p.Gln1756Profs), 300T>G (c.181T>G; p.Cys61Gly), 185delAG (c.68_69delAG; p.Glu23Valfs), and 4153delA (c.4035delA; p.Glu1346Lysfs) [10, 11].
BRCA1 mutations associated with breast cancers are usually basal-like and triple-negative without HER2 gene amplification [12]. The risk of breast cancer for BRCA1 and BRCA2 germline mutations among the carriers under 80 years of age is approximately 72% and 69%, respectively. Similarly, the risk of serous ovarian cancer is 44% for BRCA1 and 17% for BRCA2 [13, 14]. Homozygous nonfunctional mutations of BRCA1 usually result in embryo lethality. The biallelic compound heterozygous variant of BRCA1 containing c.594_597del4 and c.5095C>T is viable but exhibits susceptibility to microsomia, dysmorphia, mild intellectual disability, crosslinking agents, and breast cancer [15].
Here, we present a case report of serous ovarian cancer in a 67-year-old female patient and a novel germline mutation that has not been yet described either in Polish patients or in any other population.
Materials and methods
Study subjects
The examined material included blood cells (BC) and formalin-fixed paraffin-embedded tissue (FFPE) collected from a proband during standard laboratory procedures. The histological diagnosis of ovarian serous carcinoma and the tumor tissue was evaluated by a pathologist. Blood samples were collected from the proband's daughter, who had breast cancer in 2017, and from the proband's healthy son.
The patient, a 67-year-old female, underwent right-sided mastectomy for breast cancer (NST, G2, T2N0M0) in 2011 and complementary chemotherapy (FEC) in 2012. Furthermore, she suffered from hypogastric pain in July 2017, while ascites and tumors of the left and right adnexa were detected in September of the same year. The level of CA125 was 14749 U/ml. The use of standard tests did not reveal the presence of founder mutations in BRCA1/BRCA2 characteristic of the Polish population.
The patient was treated with chemotherapy: 6 courses of Carboplatin AUC5 and Paclitaxel, which brought significant regression of the symptoms. In April 2018, the patient underwent radical surgery i.e. bilateral oophorectomy and hysterectomy. Diagnosis: G2 ovarian serous cancer with metastases to the abdomen. In November 2018, disease progression was recorded. In January 2019, the patient was reoperated on, and second-line chemotherapy was implemented: 6 courses of Carboplatin and Paclitaxel with complete response in imaging and marker tests. BRCA1/2 mutation tests in archival material were ordered. A new, probably pathogenic BRCA1 gene variation was found using the NGS technology. The patient was qualified for maintenance therapy with Olaparib (800 mg daily).
Approval for this study was obtained from the Bioethical Commission in Lublin (approval number 10/2020/KB/VIII). All procedures were performed in accordance with the Declaration of Helsinki (ethical principles for medical research involving human subjects).
DNA Extraction and Genotyping
Genomic DNA was extracted from the whole blood using a commercial kit NucleoSpin Dx Blood (Machery-Nagel, Duren, Germany) according to the manufacturer's instructions. The quantity and quality of DNA were determined with the use of a BioPhotometer (Eppendorf, Hamburg, Germany). Three mutations of the BRCA1 gene: 5382insC (c.5266dupC), 300T>G (c.181T>G), and 4153delA (c.4035delA) were amplified by PCR using primers and conditions described by [16],[17]. For other mutations: 185delAG (c.68_69delAG; p.Glu23Valfs), 3819delGTAAA (c.3700_3704delGTAAA; (p.Val1234Glnfs), 3875delGTCT (c.3756_3759delGTCT; p.Ser1253Argfs), 3896delT (c.3779delT; p.Leu1260Tyrfs), 4160delAG (c.4041_4042delAG, p.Gly1348Asnfs), and 4184delTCAA (c.4065_4068delTCAA; p.Asn1355Lysfs), primers were designed, and reaction conditions were experimentally determined (Table S1).
Primers were designed using the freely available Software Primer [18]. The Taq PCR Master Mix kit (EURx, Gdansk, Poland) was used to prepare the PCR reaction (with 40 - 60 ng DNA) in a SimplyAmpTM thermal cycler (Applied Biosystems, Thermo Fisher Scientific). The PCR products were enzymatically purified using an ExoBap kit (EURx, Gdansk, Poland). The sequential PCR was then prepared using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). In the next step, the terminators were removed after PCR using ExTerminator (A&A Biotechnology, Gdynia, Poland). After the purification, all samples were placed on a 96-well plate and sequenced using a 3100 Capillary Sequencer (Applied Biosystems, Thermo Fisher Scientific). Next, the results were pre-analyzed by ABI3100 Data Collection Software. Then, the data files of the samples were checked by FinchTV1.4 (Geospiza) and compared with reference sequences BRCA1: NM_007294.3.
Multiplex Ligation-Dependent Probe Amplification (MLPA) Analysis
BRCA1/2 genomic arrangements were searched with the MLPA method [19] according to the manufacturer's instructions (MRC-Holland, Amsterdam, the Netherlands) using P087 SALSA and P090 SALSA MPLA kits. The products of MPLA reactions were diluted 10 times in Hi-DiTM Formamide (Applied Biosystems), and GeneScanTM 500 ROXTM dye Size Standard (Applied Biosystems) was added to each sample (as an internal lane size standard). It facilitated automated data analysis. Moreover, it was essential for the achievement of high run-to-run precision in sizing DNA fragments by electrophoresis. The samples were size-separated by capillary electrophoresis (POP7 polymer, ABI PRISM 3100, Applied Biosystems). The electropherograms were analyzed by GeneMarker software (version 2.2.0, SoftGenetics, State College, PA, USA).
NGS Sequencing
The number of cancer cells (evaluated by the pathologist) was approximately 100% in the FFPE sample. DNA was extracted using GeneRead DNA FFPE (QIAGEN, Hilden, Germany). The DNA concentration was determined using a Qubit I fluorometer and a dsDNA High Sensitivity Assay Kit (InvitrogenTM, ThermoFisher Scientific).
The DNA was diluted to a concentration of 10 ng/µl. The libraries were prepared using a Devyser BRCA kit (Hagersten, Sweden) according to the manufacturer's instructions. Complete sequence determination of all coding exons and all adjacent exon-intron boundaries (minimum 20 bp proximal to 5' end and 10 bp distal to 3' end of each exon) were covered. First, amplicon libraries were generated in one multiplex PCR reaction (PCR1). Next, index addition to PCR1 was performed in PCR2. The sample libraries obtained in PCR2 were pooled and purified in a single tube. The library concentration was quantified using a Qubit I fluorometer and a dsDNA High-Sensitivity Assay Kit (InvitrogenTM, Thermo Fisher Scientific). The purified library pool was diluted to a 0.4 ng/ml concentration and denatured.
Sequencing was performed on MiniSeq (Illumina, San Diego, USA) using Mid Output Reagent Cartridge 300 cycles (Illumina, San Diego, USA). The Devyser BRCA libraries were sequenced in a paired-end mode (2 x 151 bp). The results of sequencing data files (FASTQ) were reviewed using Amplicon Suite (Smartseq s.r.l).
Data Analysis
All mutations were reported following the Human Genome Variation Society (HGVS) guidelines (http://varnomen.hgvs.org/) based on the coding sequences: NM_007294.3 for BRCA1 and NM_000059.3 for BRCA2. For the detection of sequence variants of somatic origin, we ensured that each amplicon had at least 1,000 coverage for detection of variant allele frequency (VAF) down to 5%. Mutations were classified according to clinical variants (https://www.ncbi.nlm.nih.gov/clinvar/), mutations in the COSMIC database (https://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), variants of unknown significance, literature search, and in silico analyses using Varsome (https://varsome.com/). The presence of BRCA1/2 genes mutations was confirmed by Sanger sequencing (Table S1).
Results
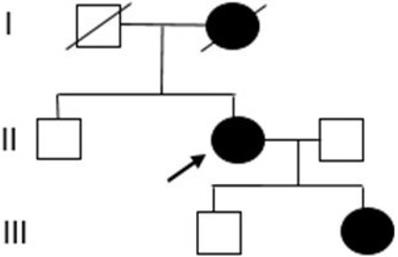
The proband was referred to genetic counseling since her mother was diagnosed with breast cancer (Figure 1).
Pedigree of the proband's family. (I) mother with breast cancer, (II) proband with breast and ovarian cancer, and (III) daughter with breast cancer.
Nine mutations in BRCA1 were genotyped, i.e., c.5266dupC (p.Gln1756Profs), c.181T>G (p.Cys61Gly), c.68_69delAG (p.Glu23Valfs), c.4035delA (p.Glu1346Lysfs), c.3700_3704delGTAAA (p.Val1234Glnfs), c.3756_3759delGTCT (p.Ser1253Argfs), c.3779delT (p.Leu1260Tyrfs), c.4041_4042delAG (p.Gly1348Asnfs), and c.4065_4068delTCAA (p.Asn1355Lysfs), and were not detected in the samples from the proband.
MLPA analysis was used to test the presence of large genomic rearrangements (LGRs) in BRCA1 and BRCA2. No deletions or duplications of genomic DNA fragments, i.e., copy number variations (CNV), were detected in the patient and patient's daughter samples (Figures S1A, S1B, S1C, S1D).
The data coverage for the FFPE sample showed a mean amplicon reading depth per sample ranging from 9813 to 2690 for BRCA1 and from 17980 to 3237 for BRCA2. Seven single nucleotide variants (SNVs) and one deletion were identified in the BRCA1 gene by NGS, whereas five SNVs and one deletion were detected in BRCA2 (Table 1).
BRCA1 and BRCA2 variants detected by NGS
| c.DNA | Protein | Type | RS number | Classification | % reads |
|---|---|---|---|---|---|
| BRCA1 | |||||
| c.2612C>T | p.Pro871Leu | SNV | rs799917 | benign [class 1] | 5.96 |
| c.4308T>C | p.Ser1436= | SNV | rs1060915 | benign [class 1] | 5.58 |
| c.3113A>G | p.Glu1038Gly | SNV | rs16941 | benign [class 1] | 5.85 |
| c.2311T>C | p.Leu771= | SNV | rs16940 | benign [class 1] | 5.94 |
| c.2082C>T | p.Ser694= | SNV | rs1799949 | benign [class 1] | 6.23 |
| c.3548A>G | p.Lys1183Arg | SNV | rs16942 | benign [class 1] | 6.63 |
| c.4837A>G | p.Ser1613Gly | SNV | rs1799966 | benign [class 1] | 5.69 |
| c.829_832delAATA | p.Asn277Leufs20 | del | Pathogenic [class 5] | 94.75 | |
| BRCA2 | |||||
| c.68-7delT | del | rs276174878 | Uncertain significance [class 3] | 16.58 | |
| c.6513G>C | p.Val2171= | SNV | rs206076 | benign [class 1] | 99.29 |
| c.4563A>G | p.Leu1521= | SNV | rs206075 | benign [class 1] | 99.24 |
| c.7806-14T>C | SNV | rs9534262 | benign [class 1] | 50.87 | |
| c.7397T>C | p.Val2466Ala | SNV | rs169547 | benign [class 1] | 99.4 |
| c.3807T>C | p.Val1269= | SNV | rs543304 | benign [class 1] | 50.93 |
All detected SNVs of BRCA1 were reported in ClinVar as benign (class 1). In this case, one probably pathogenic variant in BRCA1 was identified: c.829-832delAATA, p.Asn277Leufs*20, NM_007294. This genetic variant is a frame-shift mutation resulting in a premature stop codon and protein truncation. This mutation is located in exon 10 of 23 (159 - 162 of 3426 codings, NMD).
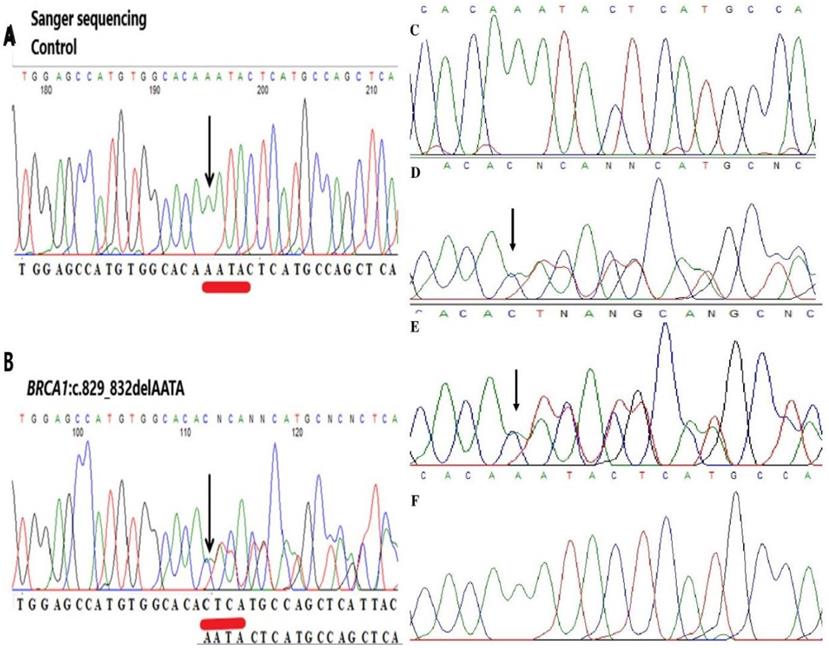
To our knowledge, this mutation has not been reported yet in the Polish population and elsewhere. Additionally, this variant was fully confirmed by Sanger sequencing; therefore, the estimated specificity was 100% (Figure 2). A new variant was detected in the proband and the proband's daughter. This change was not detected in the son's samples.
Representative image of validation of NGS data by Sanger sequencing. (A, C) Sequencing analysis of genomic DNA from the control. (B, D) Sequencing analysis of genomic DNA from the proband carrying the c.829_832delAATA mutation in the BRCA1 gene. (E) Sequencing analysis of genomic DNA from the daughter carrying the c.829_832delAATA mutation in the BRCA1 gene. (F) Sequencing analysis of genomic DNA from the son in the BRCA1 gene. The arrows indicate the position of the mutated nucleotides.
Five detected BRCA2 SNVs were reported as benign (class 1). One variant c.68-7delT was reported as a conflicting interpretation of pathogenicity (class 3) (Table 1).
Discussion
Determination of the status of mutations in the BRCA1/2 genes is important for the identification of a predisposition to familial cancer and for the selection of proper therapy. Hereditary mutations in the BRCA1 and BRCA2 genes are risk factors for ovarian cancer. Especially drugs based on PARP inhibitors and platinum can be used in patients with such mutations. New techniques can be used for the analysis of not only germline mutations but also somatic gene variants.
In BRCA1, one deletion c.829_832delAATA was detected by NGS. According to the American College of Medical Genetics and Genomics (ACMG) guidelines [20, 21], this variant may be classified as pathogenic, fulfilling the PVS1 criteria (pathogenic, very strong), PM2 (pathogenic moderate), or PP3 (supporting), and associated with breast and ovarian cancer. The pathogenic PVS1 criterion means a null variant (frame-shift), which is a known mechanism of disease. The BRCA1 gene has about 2,908 known pathogenic variants (which is greater than the minimum of 0.7) associated with breast-ovarian cancer, familial, susceptibility to, 1, pancreatic cancer, susceptibility to, 4 and Fanconi anemia, complementation group S [20, 21].
The PM2 criterion means absent in controls (or at an extremely low frequency if recessive) in the Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium. Variants not found in GenomAD exomes coverage = 62.4 and not found in GnomAD genomes (good GnomAD genomes coverage = 32.2). PP3 pathogenic verdict computational was defined on 1 prognosis (GERP) vs. no benign. The germline BRCA1 mutation c.829_832delAATA was confirmed as well (Figure 1).
The NGS analysis of BRCA2 in the FFPE sample showed five SNV-type variations and one deletion. The c.68_7delT variant was not precisely qualified according to the ClinVar database. Two of the four annotations describe it as a variant of uncertain significance (VUS) (class 3) according to the ACMG guidelines [20], and the other two as benign (class 1) variants [21]. The https://varsome.com/ database based on the American College of Medical Genetics and Genomics Classification indicates this variant as a change of unknown significance (class 3) [21].
In this case, no mutation previously described as a Polish-founder mutation was identified. The NGS-based testing allowed optimization of the detection of new variants of the BRCA1/2 genes.
The use of PARP inhibitors in targeted therapy is beneficial for patients with BRCA1/2 mutations [22, 23, 24]. Parp1 plays a key role in the repair of single-strand DNA breaks [25]. Recent studies indicate that NGS is increasingly being used in routine testing by laboratories. The use of this technology brings benefits to patients in terms of analysis time. NGS is recognized as an efficient method in the detection of both inheritable and acquired mutations using DNA from FFPE [26].
Conclusions
In our work, we demonstrated that the use of standard procedures may not be sufficient in the diagnosis of hereditary and somatic BRCA1/BRCA2 mutations. We have shown that NGS with a commercial kit for detection of BRCA1/2 mutants from FFPE tumor tissue is fully efficient. Quick determination of pathogenic variants is important to facilitate specific therapy, in addition to the identification of familial predispositions to cancer. Discovering new variants that are inherited and that can cause familial cancers will facilitate monitoring other family members. Such an approach may help in the faster detection of undesirable changes that may cause cancer.
Abbreviations
ACMG: American College of Medical Genetics and Genomics; BC: Blood Cells; BRCA1: Breast Cancer 1, Early Onset; BRCA2: Breast Cancer 2, Early Onset; CNV: Copy Number Variants; FFPE: Formalin-Fixed Paraffin-Embedded; HGVS: Human Genome Variation Society; LGR: Large Genetics Rearrangement; MLPA: Multiplex Ligation-Dependent Probe Amplification; NGS: Next Generation Sequencing; PARP: Poly (Adenosine Diphosphate)-Ribose Polymerase; SNV: Single Nucleotide Variant; VAF: Variant Allele Frequency; VUS: Variant as of Uncertain Significance.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We thank all patients who agreed to participate in the current study.
Author Contributions
MO, JW participated in the research design, analyzed and interpreted the data, and drafted the manuscript; JP, JSS, KOB provided samples and compiled clinical data. All authors have read and approved the final version of the manuscript.
Availability of data and materials
The data that support the findings of this study are not publicly available to protect patient privacy, but they are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
All procedures were approved by the Bioethical Commission in Lublin (approval number 10/2020/KB/VIII). All data were fully anonymized prior to access by the authors.
All procedures were performed in accordance with the Declaration of Helsinki (ethical principles for medical research involving human subjects). Written informed consent for participation in the study and publication was obtained from the patient described in this case report.
Consent for publication
The proband provided a signed consent form prior to molecular genetic testing for the permission of the anonymous use of her data for research purposes and/or scientific publications.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941-53
3. Ratajska M, Krygier M, Stukan M, Kuźniacka A, Koczkowska M, Dudziak M. et al. Mutational analysis of BRCA1/2 in a group of 134 consecutive ovarian cancer patients. Novel and recurrent BRCA1/2 alterations detected by next generation sequencing. J Appl Genet. 2015;56:193-198
4. Ellison G, Ahdesmäki M, Luke S, Waring PM, Wallace A, Wright R. et al. An evaluation of the challenges to developing tumor BRCA1 and BRCA2 testing methodologies for clinical practice. Hum Mutat. 2018;39:394-405
5. Jasin M, Rothstein R. Repair of Strand Breaks by Homologous Recombination. Cold Spring Harb Perspect Biol. 2013 5:(11) a012740
6. Shabbeer S, Omer D, Berneman D, Weitzman O, Alpaugh A, Pietraszkiewicz A. et al. BRCA1 targets G2/M cell cycle proteins for ubiquitination and proteasomal degradation. Oncogene 2013; 32:42. 2012;32:5005-16
7. Naseem R, Sturdy A, Finch D, Jowitt T, Webb M. Mapping and conformational characterization of the DNA-binding region of the breast cancer susceptibility protein BRCA1. Biochem J. 2006;395:529-35
8. Matsuzawa A, Kanno S ichiro, Nakayama M, Mochiduki H, Wei L, Shimaoka T. et al. The BRCA1/BARD1-interacting protein OLA1 functions in centrosome regulation. Mol Cell. 2014;53:101-14
9. Paul A, Paul S. The breast cancer susceptibility genes (BRCA) in breast and ovarian cancers. Front Biosci (Landmark edition). 2014;19:605-18
10. Górski B, Byrski T, Huzarski T, Jakubowska A, Menkiszak J, Gronwald J. et al. Founder mutations in the BRCA1 gene in Polish families with breast-ovarian cancer. Am J Hum Genet. 2000;66:1963-8
11. Kowalik A, Siołek M, Kopczyński J, Krawiec K, Kalisz J, Zięba S. et al. BRCA1 founder mutations and beyond in the Polish population: A single-institution BRCA1/2 next-generation sequencing study. PloS one. 2018;13(7):e0201086
12. Jönsson G, Staaf J, Vallon-Christersson J, Ringnér M, Holm K, Hegardt C. et al. Genomic subtypes of breast cancer identified by array-comparative genomic hybridization display distinct molecular and clinical characteristics. Breast Cancer Res: BCR. 2010;12:R42
13. KB K, JL H, DR B, KA P, TM M, MJ R-B. et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA. 2017;317:2402-16
14. Lubinski J, Huzarski T, Gronwald J, Cybulski C, Debniak T, Sun P. et al. Age-specific risks of incident, contralateral and ipsilateral breast cancer among 1776 Polish BRCA1 mutation carriers. Breast Cancer Res Treat. 2019;174:769-74
15. Sawyer SL, Tian L, Kähkönen M, Schwartzentruber J, Kircher M, Majewski J. et al. Biallelic Mutations in BRCA1 Cause a New Fanconi Anemia Subtype. Cancer Discov. 2015;5:135-42
16. Friedman LS, Ostermeyer EA, Szabo CI, Dowd P, Lynch ED, Rowell SE. et al. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet. 1994;8:399-404
17. Górski B, Jakubowska A, Huzarski T, Byrski T, Gronwald J, Grzybowska E. et al. A high proportion of founder BRCA1 mutations in Polish breast cancer families. Int J Cancer. 2004;110:683-6
18. Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Mol Biol. (Clifton, NJ). 2000;132:365-86
19. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57
20. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24
21. Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY. et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017 19:10. 2017;19:1105-17
22. Tewari KS, Eskander RN, Monk BJ. Development of Olaparib for BRCA-Deficient Recurrent Epithelial Ovarian Cancer. Clin Cancer Res. 2015;21:3829-35
23. Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A. et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949-61
24. Oza AM, Matulonis UA, Malander S, Hudgens S, Sehouli J, del Campo JM. et al. Quality of life in patients with recurrent ovarian cancer treated with niraparib versus placebo (ENGOT-OV16/NOVA): results from a double-blind, phase 3, randomised controlled trial. Lancet Oncol. 2018;19:1117-25
25. Rodriguez-Freixinos V, Fariñas-Madrid L, Gil-Martin M, Barretina-Ginesta P, Romeo M, Villacampa G. et al. Chemotherapy and PARP inhibitors in heavily pretreated BRCA1/2 mutation ovarian cancer (BMOC) patients. Gynecol Oncol. 2019;152:270-7
26. Mafficini A, Simbolo M, Parisi A, Rusev B, Luchini C, Cataldo I. et al. BRCA somatic and germline mutation detection in paraffin embedded ovarian cancers by next-generation sequencing. Oncotarget. 2016;7:1076-83
Author contact
![]() Corresponding author: Malgorzata Ostrowska Department of Biotechnology, Microbiology and Human Nutrition, University of Life Sciences in Lublin, 8 Skromna St., 20-704 Lublin, Poland, tel: 48 81 46 23 402, E-mail: malgorzata.ostrowskalublin.pl
Corresponding author: Malgorzata Ostrowska Department of Biotechnology, Microbiology and Human Nutrition, University of Life Sciences in Lublin, 8 Skromna St., 20-704 Lublin, Poland, tel: 48 81 46 23 402, E-mail: malgorzata.ostrowskalublin.pl