ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2021; 9:38-42. doi:10.7150/jgen.58823 This volume Cite
Research Paper
Genetic Diversity of Clinical Bordetella Pertussis ST2 Strains in comparison with Vaccine Reference Strains of India
Naresh Chand Sharma1, Shalini Anandan2, Naveen Kumar Devanga Ragupathi2,3, Dhiviya Prabaa Muthuirulandi Sethuvel2, Karthick Vasudevan2, Dhirendra Kumar1, Sushil Kumar Gupta1, Lucky Sangal4, Balaji Veeraraghavan2 ![]()
1. Maharishi Valmiki Infectious Diseases Hospital, New Delhi - 110009, India.
2. Department of Clinical Microbiology, Christian Medical College, Vellore - 632 004, India.
3. Department of Chemical and Biological Engineering, The University of Sheffield, Sheffield, United Kingdom.
4. World Health Organization, Country Office, New Delhi - 110029, India.
Received 2021-1-30; Accepted 2021-5-11; Published 2021-9-3
Abstract

Objectives: Pertussis is a highly contagious disease of the respiratory tract caused by Bordetella pertussis, a bacterium that lives in the mouth, nose, and throat. Current study reports the highly accurate complete genomes of two clinical B. pertussis strains from India for the first time.
Methods: Complete genome sequencing was performed for two B. pertussis strains using Ion Torrent PGM and Oxford nanopore sequencing method. Data was assembled de novo and the sequence annotation was performed through PATRIC and NCBI server. Downstream analyses of the isolates were performed using CGE server databases for antimicrobial resistance genes, plasmids, and sequence types. The phylogenetic analysis was performed using Roary.
Results: The analysis revealed insertional elements flanked by IS481, which has been previously regarded as the important component for bacterial evolution. The two B. pertussis clinical strains exhibited diversity through genome degradation when compared to whole-cell vaccine reference strains of India. These isolates harboured multiple genetic virulence traits and toxin subunits, which belonged to sequence type ST2.
Conclusion: The genome information of Indian clinical B. pertussis strains will serve as a baseline data to decipher more information on the genome evolution, virulence factors and their role in pathogenesis for effective vaccine strategies.
Keywords: Bordetella pertussis, ptx, IS481, genome reduction, ST2, Hybrid assembly
Introduction
Pertussis, caused by Bordetella pertussis, is a highly contagious respiratory infection, characterized by severe episodes of coughing and a prolonged convalescent period when the patient can transmit the disease [1]. Although large scale vaccination reduced the incidence of the disease, recent trends suggest a re-emergence particularly among the adolescent and young adult population in the developed countries. The reason for this resurgence is uncertain, apart from waning immunity which needs to be studied further. Further, the impact of vaccination needs to be established since more than one strain used in the manufacturing of pertussis vaccines globally. The comparative genomic analysis would provide high resolution data to study the structural variations in the circulating strains of B. pertussis against vaccine strains.
However, genome data of B. pertussis is rare in India due to the complexity of culturing B. pertussis from clinical samples. Recent studies revealed the genomic structural heterogeneity among the isolates within the geographical or temporally defined epidemics [2]. These studies illustrated that genome evolution in B. pertussis is mainly due to rearrangement in addition to genome reduction [2]. Genomic variations are mostly studied in the circulating strains of B. pertussis to know the pathogen adaptation to the vaccine antigens such as pertussis toxin (ptx), pertactin (prn), fimbriae (fim) and filamentous hemagglutinin (FHA) [3]. Understanding the molecular epidemiology of this pathogen is essential which will be helpful for facilitating public heath surveillance in the country. Here we report the comparative genomic analysis of two clinical isolates of B. pertussis from India against the vaccine reference strains 6229, 25525, 134, 509, 10536 and Tohama I.
Materials and Methods
Strain Isolation and Characterization
Bacterial strains BPD1 and BPD2 isolated from nasopharyngeal swabs of paediatric patients in the year 2017 as a part of surveillance study funded by World Health Organization, India. Swabs were plated in charcoal blood agar and incubated at 37 °C with CO2 for 48 hours. Isolates were confirmed by standard biochemical tests and real-time PCR for the targets IS481 and ptxS1 genes [4].
Genome sequencing
Short read sequencing and assembly
Genomic DNA of the B. pertussis isolates were extracted using QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). Whole genome sequencing (WGS) was performed in IonTorrentTM Personal Genome MachineTM (PGM) (Life Technologies, Carlsbad, CA) with 400-bp read chemistry as per manufacturer's instructions. Raw reads were assembled de novo using Assembler SPAdes v.5.0.0.0 embedded in Torrent Suite Server v.5.0.3.
Long read sequencing and assembly
Library preparation and sequencing of the B. pertussis isolates was done using SQK-LSK108 Kit R9 version (Oxford Nanopore Technologies, Oxford, UK) using 1D sequencing method according to manufacturer's protocol. Sequencing of the isolates was performed using FLO-MIN106 R9 flow cell in MinION Mk 1B sequencer. Briefly, genomic DNA was purified using 1X AMPure beads (Agencourt, Beckman Coulter, Brea CA, USA). Purified DNA was subjected to dA-tailing using NEBNext dA-Tailing Module (New England Biolabs), followed by ligation with the leader and hairpin sequencing adapters (Oxford Nanopore Technologies, Oxford, UK) using Blunt TA Ligase master mix (New England Biolabs). Barcode adapters were ligated using BAM 1D (Oxford Nanopore Technologies) followed by AMPure beads purification and elution in 15 μL of Elution Buffer (Oxford Nanopore Technologies, Oxford, UK). Prepared DNA was then loaded onto the flow cell.
To perform sequencing, MinKNOW software ver. 1.15.1 (Oxford Nanopore Technologies, Oxford, UK) was used in a Windows platform and raw data (fast5 files) were obtained. The Fast5 files were basecalled with Albacore 2.0.1 (https://nanoporetech.com/about-us/news/new-basecaller-now-performs-raw-basecalling-improved-sequencing-accuracy). Error correction and genome assembly was performed using Canu 1.7 [5]. The obtained contigs were polished with Nanopolish 0.10.1 (https://github.com/jts/nanopolish) after de novo assembly.
Hybrid assembly using IonTorrent and MinION reads
Hybrid assembly using both IonTorrent and MinION reads were performed to increase the accuracy and completeness of genome. Unicycler (v0.4.6) was used for generating hybrid assemblies [6]. Further, the reads were polished with multiple rounds of Pilon v1.24 [7] to reduce the base level errors. The assembly statistics and average nucleotide identity of different assemblies were evaluated using Quast v5.0.2 [8].
Genome annotation and MLST analysis
Annotation of the sequences were done using PATRIC, the bacterial bioinformatics database and analysis resource (http://www.patricbrc.org) [9], and NCBI Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP, http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html). Taxonomy identification was performed using PathogenFinder 1.1 (https://cge.cbs.dtu.dk/services/PathogenFinder/).
Virulence genes and antibiotic resistance genes were identified using VFDB database (http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi) and ResFinder 4.0 (https://cge.cbs.dtu.dk/services/ResFinder/). Mobile genetic elements (MGEs) like prophages, CRISPR, and Genomic islands were analysed using PHASTER, CRISPR Finder, and Island viewer [10-12]. Plasmids were identified using PlasmidFinder 2.1 (https://cge.cbs.dtu.dk/services/PlasmidFinder/). MLST 1.8 (Multi Locus Sequence Typing) tool was employed for sequence type analysis (https://cge.cbs.dtu.dk//services/MLST/) [13].
Phylogenomic analysis
The clinical strains BPD1 and BPD2 were compared with the previously reported vaccine reference strains 6229 (CP017404), 25525 (CP017405), 134 (CP017402), 509 (CP017403), 10536 (CP012228) and Tohama I (NC_002929). The phylogenetic analysis was performed using Roary: The Pan Genome Pipeline from Sanger Institute v3.11.2 [14]. Phylogenetic tree was constructed using Interactive Tree of Life (iTOL) v3 [15].
Results and Discussion
Genome length, CDS and ST types
Hybrid assemblies returned with 203X and 195X coverage for BPD1 and BPD2 isolates respectively for the complete genomes in MinION platform. The completed BPD1 genome had a sequence length of 4,126,211 bp with 3941 CDS, 3 rRNA and 50 tRNA, and isolate BPD2 had 4,104,911 bp with 3921 CDS, 3 rRNA and 51 tRNA (https://www.patricbrc.org). The isolates did not return any antimicrobial resistance genes or plasmids as analysed by ResFinder and PlasmidFinder. The whole genome MLST revealed the sequence type (ST) of both isolates to be ST2 belonging to CC2, previously reported to be unique to Africa [16].
Insertional elements observed in vaccine reference strains
Previous study suggests that the adaptation of B. pertussis to the human population has been associated with considerable gene loss and gene inactivation due to insertion sequence (IS) element expansion and mutations, a process commonly seen in host-restricted bacteria [17]. In this study, BPD1 had included ~120 Kb repeat insertion flanked by copies of IS481 in single copy (Table 1), while, BPD2 had an insertion of ~180 Kb length. However, these isolates lacked other repeat regions that were observed in the vaccine reference strains 6229 (CP017404) and 25525 (CP017405) used for production of whole-cell vaccine (WCV) in India belonging to ST2 [2]. Comparison of the repeat region observed in BPD1 with 25525 reference strain using Easyfig v2.2.3 showed the similarity between the two regions with internally inverted repeat regions which is absent in BPD2 (Figure 1). In addition, three copies of same region were found in 25525 while only one copy was found in BPD1 which mainly contains flagellar genes, transcriptional regulators, and hypothetical proteins. Whereas the comparison of the BPD2 with 25525 genome exhibits the presence of a repeat region different than in the vaccine reference strains (Figure 2). These repeat regions carry flagellar genes, efflux genes, translation initiation factor, transcriptional regulators, ribosome binding protein, hypothetical proteins and genes involved in metabolism of B. pertussis. Such transposable DNA elements were regarded as the potent force in the evolution of bacteria [18].
Virulence genome characteristics of clinical B. pertussis from India
| Strain | BPD1 | BPD2 |
|---|---|---|
| Accession number | CP034182 | CP034101 |
| ~120 Kb Repeat insertion flanked by copies of IS481 1,240,892 to 1,359,872 | √ | - |
| ~180 Kb Repeat insertion flanked by copies of IS481 2,186,762 to 2,372,309 | - | √ |
| Virulence factors | ||
| Pertactin; prn | √ | √ |
| Dermonecrotic toxin | √ | √ |
| Pertussis toxin; ptxS1, ptxS2, ptxS3, ptxS4, ptxS5 | √ | √ |
| Putative toxin; Toxin subunit PtxB/PtxC-related protein | √ #, G45S | √ #, G45S |
| Bifunctional adenylate cyclase toxin/ hemolysin CyaA | √ | √, G552R |
| RTX toxins determinant A and related Ca2+-binding proteins/Cytolysin-adenylate cyclase | √ | √ |
| toxin-activating lysine-acyltransferase | √ | √ |
| toxin-antitoxin system CptAB antitoxin | √ | √ |
| type II toxin-antitoxin system HipA family toxin | √ | √ |
| type II toxin-antitoxin system HicB family antitoxin | √ | √ |
| type II toxin-antitoxin system HicA family toxin | √ | √ |
| type II toxin-antitoxin system RatA family toxin | √ | √ |
| type II toxin-antitoxin system MqsA family antitoxin | √ | √ |
| type II toxin-antitoxin system MqsR family toxin | √ | √ |
| Type III secretion proteins | √ | √ |
| Type IV secretion proteins | √ | √ |
| virB2 - B11 | √ | √ |
| Filamentous hemagglutinin fhaB, fhaC | √ *, G280S, A281T | √ |
* fhaB; # ptxB.
Representation of similarity and internal inverted repeats in comparison of repeat region flanked by IS481 from BPD1 (CP034182) and the vaccine reference strain 25525 (CP017405). Three copies of same region were found in 25525, whereas only one copy was identified in BPD1.
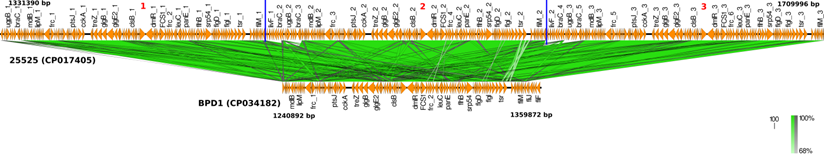
Comparison of BPD2 (CP034101) and vaccine strain 25525 (CP017405) revealed the presence of insertion with repeat region flanked by IS481 in BPD2 which is absent in the vaccine strain.
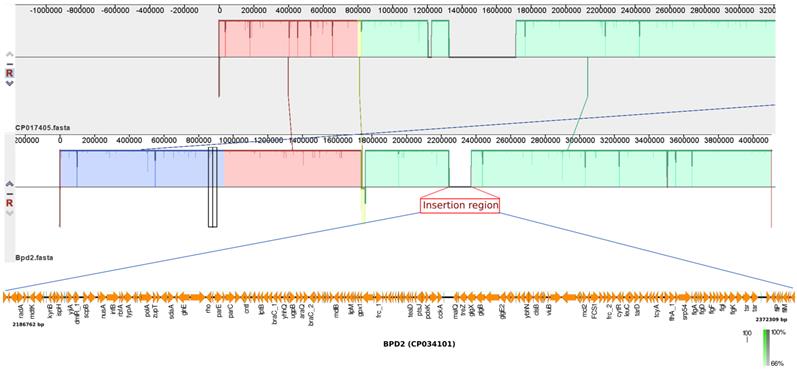
The homologous recombination between copies of IS481 has been attributed to genome reduction in B. pertussis which also suggests possible genome expansion by the same mechanism. Similarly, BPD1 and BPD2 have undergone genome reduction due to IS481 comparable to vaccine strains 6229 and 25525. The lesser number of structural genes adds up to the potential of B. pertussis to be more virulent as it reduces the number of targets that are available for recognition by the human immune system [19].
Genomic plasticity in natural populations of B. pertussis is mainly through gene acquisition, loss, and genomic organization. Horizontal gene transfer (HGT) is one of the mechanisms responsible for genome evolution in B. pertussis [20]. In this study, analysis of mobile genetic elements showed that BPD2 isolate had one intact phage region while no intact phages has been found in BPD1. Whereas genomic islands consisting of genes involved in carbohydrate, amino acid metabolism, membrane transport and transposases were identified in both the isolates. However, we did not observe CRISPR sequences, plasmids, acquired antibiotic resistance genes and mutation in 23S rRNA gene associated with macrolide resistance in B. pertussis.
Phylogenomic relation between clinical strains and vaccine reference strains
Core-genome based phylogenetic analysis of the clinical strains and vaccine reference strains revealed that 6229 and 25525 were reported to be more closely related to BPD1 and BPD2 than the other vaccine strains of India 134 (CP017402), 509 (CP017403) and 10536 (CP012228) (Figure 3) [2, 21]. This could be due to the switch between whole-cell and acellular vaccines. Interestingly, 134 was found to be closely related to the global reference strain Tohama I. Whereas 509 and 10536 showed high genetic distance to all other strains.
Core-genome based phylogenetic comparison of BPD1 and BPD2 Indian clinical strains with 134 (CP017402), 509 (CP017403), 10536 (CP012228), and Tohama I (NC_002929) vaccine reference strains.

Toxin and other virulence genes
The isolates were analysed for the presence of major virulence and toxin genes including pertactin (prn), fimbriae (fim2, fiim3), filamentous (FHA) and pertussis toxin (dnt, cyaA, ptxA, ptxB, ptxC, ptxD, ptxE, ptxS). Both BPD1 and BPD2 isolates had ptxS toxin genes with all five subunits and carried other genes with no allelic variation compared to vaccine reference strains except cyaA gene in BPD2 which had amino acid substitution of G552R and ptxB gene in both the isolates had substitution of G45S. This observation supports the hypothesis that pathogen adaptation is mainly through antigenic divergence between vaccine strains and circulating strains [17]. Other genes identified include virB2, B3, B6, B7, B8, B9, B10 and B11 (Table 1).
Pathogen identities
Moreover, the PathogenFinder for BPD1 proteome families matched with 10 non-pathogenic families and 3 pathogenic families. This showed sequence similarity of 88%, 85.9% and 87.18% to the pathogenic families of Burkholderia cenocepacia, Burkholderia mallei and Neisseria gonorrhoeae respectively. Similar results were observed for BPD2.
Conclusion
Utilizing the hybrid genome technology, we report the highly accurate complete genome sequences of B. pertussis Indian clinical strains. Comparison of clinical strains with vaccine reference strains of India revealed presence of existing and new insertional regions (~120 KB and ~180 KB) attributing to the genome degradation and expansion. Also, the resulting antigenic variation against the vaccine reference indicates that B. pertussis is evolving under vaccine driven selection. The impact of these events needs further investigation using a larger collection of clinical isolates. However, this comparative genome analysis will help to decipher more information on the genome evolution, virulence factors and their role in pathogenesis for the development of effective vaccine strategies in India.
Acknowledgements
The authors thank the funding agency, WHO Country Office for India for supporting this study. The research work has been approved by the Institutional Review Board of Christian Medical College, Vellore at the meeting conducted on 28/10/2016 (IRB Min No: 9706). NKDR is funded by the Global Challenges Research Fellowship, The University of Sheffield, UK.
Funding
This study was supported by World Health Organization, Country office, New Delhi, India. (Ref. No. 2015/561588-0).
Nucleotide accession numbers
The complete genome sequence of BPD1 and BPD2 were deposited at DDBJ/ENA/GenBank under the accession CP034182 and CP034101, respectively.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cherry JD. Historical review of pertussis and the classical vaccine. J Infect Dis. 1996;174:S259-63
2. Weigand MR, Peng Y, Loparev V. et al. Complete Genome Sequences of Four Bordetella pertussis Vaccine Reference Strains from Serum Institute of India. Genome announc. 2016 4(6). pii: e01404-16. doi: 10.1128/genomeA.01404-16
3. Alai S, Ghattargi VC, Gautam M. et al. Comparative genomics of whole-cell pertussis vaccine strains from India. BMC Genomics. 2020;21:345. https://doi.org/10.1186/s12864-020-6724-8
4. Tatti KM, Sparks KN, Boney KO. et al. A novel multi-target real-time PCR assay for the rapid diagnosis of Bordetella species in clinical specimens. J Clin Microbiol. 2011: JCM-00601.
5. Koren S, Walenz BP, Berlin K. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722-36
6. Wick RR, Judd LM, Gorrie CL. et al. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput Biol. 2017;13:e1005595
7. Walker BJ, Abeel T, Shea T. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one. 2014;9:e112963
8. Gurevich A, Saveliev V, Vyahhi N. et al. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072-5
9. Wattam AR, Abraham D, Dalay O. et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucl Acids Res. 2014;42:D581-91
10. Arndt D, Grant JR, Marcu A. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:W16-21
11. Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52-7
12. Bertelli C, Laird MR, Williams KP. et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017;45:W30-5
13. Larsen MV, Cosentino S, Rasmussen S. et al. Multilocus Sequence Typing of Total Genome Sequenced Bacteria. J Clin Micobiol. 2012;50:1355-61
14. Page AJ, Cummins CA, Hunt M. et al. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691-3
15. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):W242-5
16. van Gent M, Bart MJ, van der Heide HG. et al. SNP-based typing: a useful tool to study Bordetella pertussis populations. PLoS One. 2011;6:e20340
17. Bart MJ, Harris SR, Advani A. et al. Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. MBio. 2014 5
18. Stibitz S. IS481 and IS1002 of Bordetella pertussis Create a 6-Base-Pair Duplication upon Insertion at a Consensus Target Site. J Bacteriol. 1998;180:4963-6
19. Parkhill J, Sebaihia M, Preston A. et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nature Genet. 2003;35:32-40
20. Cummings CA, Brinig MM, Lepp PW, Van De Pas S, Relman DA. Bordetella species are distinguished by patterns of substantial gene loss and host adaptation. J Bacteriol. 2004;186:1484-92
21. Weigand MR, Peng Y, Loparev V. et al. Complete genome sequences of Bordetella pertussis vaccine reference strains 134 and 10536. Genome announc. 2016;4:e00979-16
Author contact
![]() Corresponding author: Dr. V. Balaji, Professor, Department of Clinical Microbiology, Christian Medical College, Vellore - 632 004, Tamil Nadu, India. Ph: +91 9442210555; E-mail: vbalajiac.in.
Corresponding author: Dr. V. Balaji, Professor, Department of Clinical Microbiology, Christian Medical College, Vellore - 632 004, Tamil Nadu, India. Ph: +91 9442210555; E-mail: vbalajiac.in.