ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2021; 9:26-37. doi:10.7150/jgen.53019 This volume Cite
Research Paper
Analysis of Genome Sequences of Coagulase-Negative Staphylococci Isolates from South Africa and Nigeria Highlighted Environmentally Driven Heterogeneity
Tawanda Elias Maguvu ![]() , Adegboyega Oyedele Oladipo, Cornelius Carlos Bezuidenhout
, Adegboyega Oyedele Oladipo, Cornelius Carlos Bezuidenhout
Unit for Environmental Sciences and Management: Microbiology, North-West University, Potchefstroom, South Africa. Private Bag X6001, Potchefstroom, 2520, South Africa
Received 2020-9-8; Accepted 2020-10-28; Published 2021-5-13
Abstract

Here, we report high-quality annotated draft genomes of eight coagulase-negative staphylococci (CoNS) isolates obtained from South Africa and Nigeria. We explored the prevalence of antibiotic resistance and virulence genes, their association with mobile genetic elements. The pan-genomic analysis highlighted the environmentally driven heterogeneity of the isolates. Isolates from Nigeria had at least one gene for cadmium resistance/tolerance, these genes were not detected in isolates from South Africa. In contrast, isolates from South Africa had a tetM gene, which was not detected among the isolates from Nigeria. The observed genomic heterogeneity correlates with anthropogenic activities in the area where the isolates were collected. Moreover, the isolates used in this study possess an open pan-genome, which could easily explain the environmentally driven heterogeneity.
Keywords: Antibiotic resistance, Coagulase-negative staphylococci (CoNS), environmentally driven heterogeneity, Pan-genome analysis.
Introduction
Coagulase-negative staphylococci (CoNS) species have traditionally been considered commensals, however, many CoNS species are now being recognised as potential opportunistic human pathogens. Several studies have reported CoNS species being the most frequently isolated organism from bloodstream infections in intensive care units [1, 2]. Though less virulent than the Staphylococcus aureus complex, CoNS are an important reservoir of antibiotic resistance genes and virulence factors that are transferred to closely related species such as S. aureus [3]. This results in the evolution and emergence of successful clones of methicillin-resistant S. aureus (MRSA) [3]. MRSA are multidrug resistant and are resistant to the last resort β-lactam, armamentarium [4]. As a result, costs, length of hospital stay, as well as mortality resulting from infections associated with MRSA, are unsurprisingly high [5, 6]. Besides aiding MRSA through horizontal gene transfer, several outbreaks of multidrug antibiotic-resistant CoNS have been reported [7, 8, 9]. Methicillin-resistant CoNS (MR-CoNS) have a high genetic diversity particularly in genes conferring methicillin resistance [10, 11]. Thus the knowledge about genetic characteristics and variability of the species (CoNS) is really important to assess.
Here, we report high-quality draft genomes of eight CoNS isolates from two distinct geographic areas (North-West province, South Africa, and Ile Ife, Osun State, Nigeria), with distinct selective pressures on the microbes. Elevated levels of tetracycline and other antibiotics have been detected from wastewater treatment plants in the North-West province, South Africa, suggesting the overuse of these antimicrobials [12]. A number of studies reported extreme soil and surface water contamination with heavy metals (including cadmium) in and around Ile-Ife, Osun State, Nigeria [13, 14, 15, 16]. Thus, we hypothesised that these anthropogenic activities might drive genome modifications which could be reflected in the accessory genomes. We used bioinformatics platforms to analyse the genomic features of the genomes, as well as exploring the antibiotic resistance and virulent genes associated with the isolates. We highlighted environmentally driven heterogeneity among the genomes, augmenting how anthropogenic activities are impacting the microbes in our surroundings.
Materials and methods
Sample collection
Coagulase-negative staphylococci (CoNS) isolates were sampled from wastewater treatment plants in North-West province, South Africa as well as in Ile-Ife, Osun State, Nigeria. Supplementary Table 1 provides information on the samples including the created IDs and the country of origin from which the samples were collected. Isolation and preliminary identification of the isolates were done as previously described [17]. Briefly, after filtration of the wastewater, filters were plated on blood agar and mannitol/salt agar (MSA), and incubated for 24 - 48 h at 37 °C. Growth and fermentation of mannitol on MSA, as well as haemolysis on blood agar were examined and noted in all the samples. Presumptive staphylococci isolates were identified based on morphology and biochemical tests (Gram-positive cocci formed in clumps), catalase, coagulase, and DNase tests [18]. CoNS isolates were identified by API STAPH (bioMérieux) following the manufacturer's protocols. In addition, we included clinical samples provided by the Microbiology Laboratory of Ile-Ife Seventh-Day Adventist hospital, an exact geographical location as the environmental samples from Nigeria. We expected the clinical samples to be distinct from environmental samples, however, we hypothesised that anthropogenic activities in the area (cadmium pollution) will likely be reflected in both environmental and clinical isolates.
Genomic DNA extraction and sequencing library preparation
Bacterial DNA was extracted from overnight Muller-Hinton broth cultures using the Nucleospin® Tissue extraction kit (Macherey-Nagel, Düren, Germany) following the manufacturer's protocol. Agarose gel electrophoresis and NanoDrop spectrophotometry (ND-100, NanoDrop Technologies Inc, Wilmington, DE, USA) were used to determine the integrity and the purity of the resultant DNA, respectively. Paired-end libraries were prepared from 1 ng bacterial DNA using Nextera XT DNA Sample Preparation Kit and Nextera XT Index kit (Illumina Inc., San Diego, California, USA) following the manufacturers protocol. Sequencing of all the isolates shown in Supplementary Table 1, was performed using a MiSeq 2000 Illumina platform (250bp paired-end reads).
Genome assembly, functional annotation, and downstream analysis
Raw sequence reads were quality filtered and trimmed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and Trimmomatic [19], with the average quality set at Q15. De novo assembling of the quality reads was performed using SPAdes version 3.13.0. [20, 21] with minimum contig length set to > 10 000 bp. CheckM [22] was used to assess the quality of the SPAdes assembled genomes. SPAdes assembled genomes were annotated by both Prokka [23] and RAST [24] with default settings. The annotated genomes were inserted into a species tree using FastTree version 2.1.10 [25]. To determine the identity of the species, FastANI version 0.1.2 [26] was used to estimate the Average Nucleotide Identity (ANI) of the isolates using nearest genomes from the phylogenetic tree as reference genomes. Kbase App [20], was used to compute pan-genomes and generating pan-genome circle plots.
Identification of mobile genetic elements, virulence factors, and antibiotic resistance genes
Integrated prophage regions were identified and annotated by using the online PHAge Search Tool Enhanced Release (PHASTER) [27] with default parameters. PHASTER provides the prophage region length, its GC content as well as its position on the contigs. In addition, it estimates the intactness (complete/incomplete) and the most common related phages. Plasmid identification was done using PlasmidFinder 2.1 [28] with default parameters. Virulence factors were identified using the Virulence Factors of Pathogenic Bacteria Database (VFDB) [29] using S. aureus subsp aureus as a genome for comparison. The Comprehensive Antibiotic Resistance Database (CARD) [30] was used to find the antibiotic resistance genes from the isolates using the perfect and strict hits only criteria. CRISPRCasFinder [31] was used to detect clustered regularly interspaced short palindromic repeats (CRISPR) from the genome sequences.
Results
Genomic features of the Staphylococcus isolates
Assembling of the genomes with SPAdes resulted in high quality draft genomes shown in Table 1. CheckM quality assessment of the assembled genomes showed that the isolates had an average of 97.9 ± 2.4 % completeness, having average contamination of 0.48 ± 0.43 % (Supplementary Table 2). Three of the isolates (T28, T27, and T28b) had no detectable contamination. On average, 30.25 ± 17 contigs per genome were detected and this corresponds to an average N50 of 289, 514.4 ± 220,373.6. The isolate L4 (S. haemolyticus) had the smallest genome size (2,135,828) corresponding to an N50 of 41,586. This genome had the largest number of contigs (60). In contrast, T28b (S. cohnii) had the largest genome size (2,690,294) corresponding to an N50 of 566,475, having a joint least number of contigs (12) with the isolate T28 (S. cohnii) (Table 1.) The G + C content was homogeneous in all isolates averaging 32.41 ± 0.27 %. The Prokka pipeline was used for functional annotation predicting on average 2,489.8 ± 163.7 genes. Isolate L4 had the least number of predicted genes 2,162 whereas isolate D13b had the highest number of predicted genes 2,658. On average, the number of protein-coding genes was 2,444.9 ± 173.7.
General features of the genomes including the assembly metrics.
| L4 | D13b | T6 | T20 | C33 | T28 | T28b | T27 | |
|---|---|---|---|---|---|---|---|---|
| Genome Size (bp) | 2,135,828 | 2,685,820 | 2,299,081 | 2,648,007 | 2,576,421 | 2,643,319 | 2,690,294 | 2,648,416 |
| G+C Content % | 32.78 | 32.55 | 32.64 | 31.82 | 32.39 | 32.36 | 32.37 | 32.33 |
| Number of Contigs | 60 | 22 | 49 | 36 | 37 | 12 | 12 | 14 |
| Largest Contig | 247,342 | 984,404 | 278,121 | 200,372 | 434,148 | 1,296,694 | 1,298,284 | 1,296,793 |
| N50 | 41,586 | 252,300 | 76,220 | 121,032 | 131,536 | 563,483 | 566,475 | 563,483 |
| Genes | 2,162 | 2,658 | 2,284 | 2,619 | 2,497 | 2,544 | 2,603 | 2,551 |
| Protein coding gene | 2,087 | 2,626 | 2,244 | 2,601 | 2,458 | 2,499 | 2,540 | 2,504 |
| Plasmids | 0 | 1 | 1 | 0 | 0 | 2 | 3 | 2 |
Phylogenetic relationship with other Staphylococcus isolates
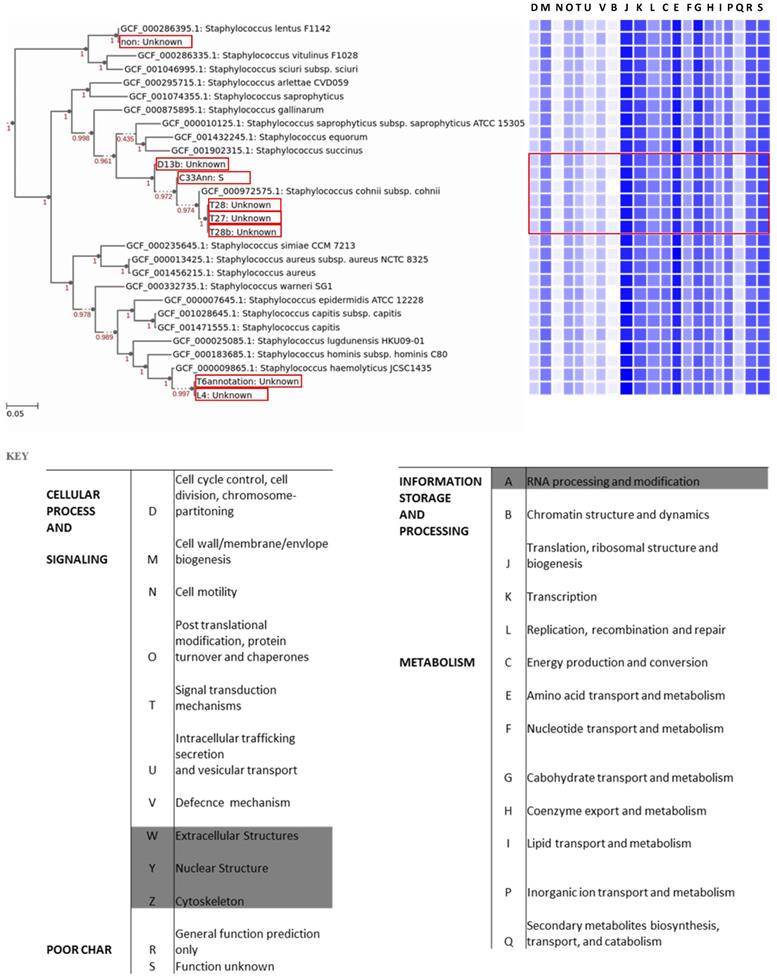
The annotated genomes were used to construct a phylogenetic tree with closely related, publicly available genomes. Core genomes of the species shown on the tree (Fig. 1) were used to determine the distribution of a selected subset of Cluster of Orthologous Groups (COGs) functional domains shown in Fig 1. The core gene category repartition was highly similar among all the isolates and the closely related species (Fig. 1). Our sequenced genomes were identified as S. lentus (T20), S. cohnii (D13b, C33, T27, T28, and T28b), and S. haemolyticus (T6 and L4) (Fig. 1). To validate the assigned species of the isolates, FastANI was used to compute the Average Nucleotide Identity (ANI) of orthologous gene pairs between the query and reference genomes. Same species are considered to have an ANI of > = 95% [27]. Staphylococcus lentus F1142 (GCF_000286395.1) when used as a reference genome had an estimated ANI of 99.1546 % with the query T20 (Table 2). They also had an approximately similar genome size of 2.5 Mb (Supplementary Fig. 1). Reference genome S. cohnii subsp. cohnii (GCF_000972575.1_assembly) had an estimated ANI of at least 98.7 % with any of (T28, T28b, T27, C33, and D13b) as a query (Table 2). Moreover, they had approximately the same genome size of 2.5 Mb (Supplementary Fig. 1). Reference genome S. haemolyticus JCSC1435 (GCF_000009865.1_assembly) had an estimated ANI of at least 98.8% with any of (T6 and L4) as the query. However, T6 and L4 had an estimated smaller genome size of 2 Mb in comparison with the reference genome size of 2.5 Mb (Supplementary Fig. 1).
Distribution of the Cluster of Orthologous Groups (COGs) function among the isolates and closely related species. The COGs were generated from the tree's core genes, annotation of the genomes was performed using RAST. Isolates from this study are highlighted in red; from top to bottom the isolates are T20, D13b, C33, T28, T27, T28b, T6, and L4, respectively. Letters D-S on top of the heat map which corresponds to letters D-S on the key represents COG categories shown on the key.
Average Nucleotide Identity (ANI) of the isolates.
| Query Genome | Reference Genome | ANI estimate | Total orthologous sequence fragments matched | Total sequence fragments of query |
|---|---|---|---|---|
| T28 | GCF_000972575.1_assembly | 99.1921 | 816 | 874 |
| (S. cohnii subsp. cohnii) | ||||
| T20 | GCF_000286395.1_assembly | 99.1546 | 743 | 863 |
| (S. lentus F1142) | ||||
| T27 | GCF_000972575.1_assembly | 99.1311 | 812 | 874 |
| (S. cohnii subsp. cohnii) | ||||
| T28b | GCF_000972575.1_assembly | 99.0809 | 821 | 890 |
| (S. cohnii subsp. cohnii) | ||||
| C33 | GCF_000972575.1_assembly | 98.9739 | 795 | 838 |
| (S. cohnii subsp. cohnii) | ||||
| T6 | GCF_000009865.1_assembly | 98.8268 | 718 | 744 |
| (S. haemolyticus JCSC1435) | ||||
| L4 | GCF_000009865.1_assembly | 98.7875 | 656 | 685 |
| (S. haemolyticus JCSC1435) | ||||
| D13b | GCF_000972575.1_assembly | 98.7468 | 805 | 884 |
Pan-Genome Analysis
The pan-genome analysis allows for better identification of diversity and composition of gene repertoire within species. The pan-genome is divided into three: (1) the core genome; which is a set of all genes common to all strains of the study, (2) non-core/accessory genome; which is a set of genes present in more than one, but not in all of the strains used in a study, and (3) singletons; which are genes unique to individual strains used in the study. A total of five isolates (D13b, C33, T27, T28, and T28b) were identified as S. cohnii, thus to have a higher resolution of the variability among these genomes, we applied the pan-genome analysis approach. Isolates (D13b and C33) had 240 and 180 singletons, respectively. In contrast, isolates (T27, T28 and T28b) had 4, 5, and 23 singletons respectively. Fig. 2 depicts the output from pan-genome analysis of the isolates identified as S. cohnii. The singletons for the isolate D13b were composed of bacteriophages, phage related genes, restriction-modification (RM) proteins and genes (Type I, II, III restriction-modification system), metal tolerance and resistance as well as stress response (Sensor protein DegS, respiratory nitrate reductase gamma chain, nitrate reductase, Multicopper oxidase, cadmium efflux system accessory protein, cadmium resistance protein, cadmium-transporting ATPase among others), several virulence determinants (Integrase, superantigen-encoding pathogenicity islands SaPI, Putative EsaC protein analog Listeria type 3), and a substantial amount of the singletons were predicted genes of unknown functions as well as hypothetical proteins. The functional distribution of the core genome is shown in Fig. 1, and it consisted of genes involved in metabolism, cellular process and signalling, information storage and signalling as well as poorly characterised functions.
Pan-genome circle plots of the S. cohnii isolates used in this study. (A) Isolate C33 is the base genome being compared with the genomes of isolates (T27, T28, T28b, and D13b). (B) Isolate D13b is the base genome being compared with genomes of isolates (C33, T27, T28, and T28b). (C) Isolate T28 is the base genome being compared with genomes of isolates (D13b, C33, T27, and T28b). (D) Isolate T27 is the base genome being compared with genomes of isolates (T28, T28b, C33, and D13b). 0 depicts the base genome on all the plots.
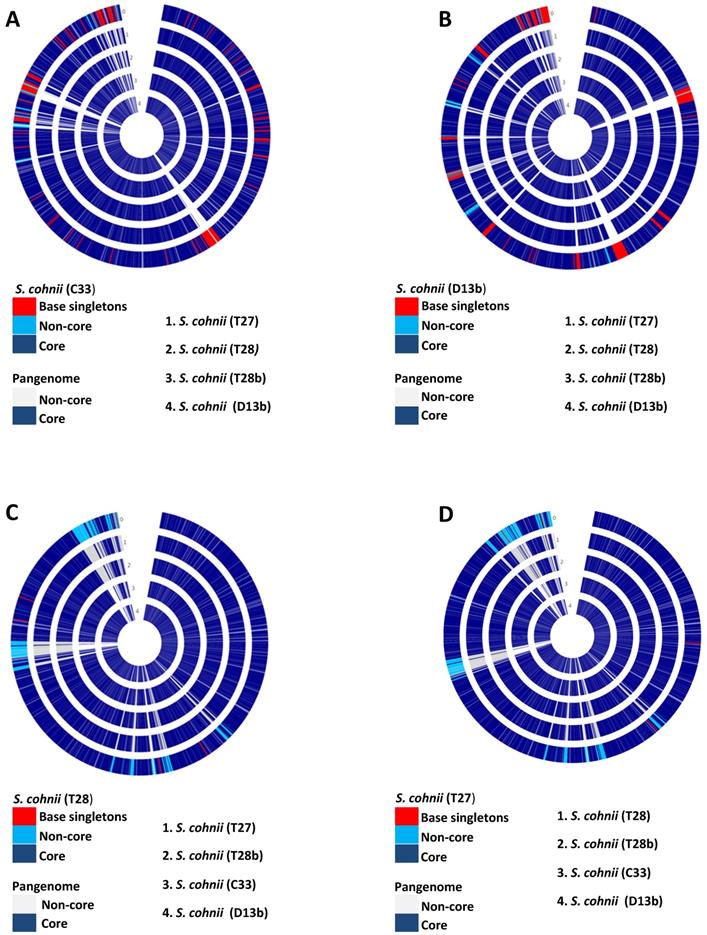
In isolate C33, the singletons covered almost similar categories to D13b, which are resistance and tolerance to stress, virulent and pathogenicity determinants with a substantial amount of hypothetical proteins, and genes of unknown functions. Some of the genes worth mentioning are Multidrug and toxin extrusion (MATE) family efflux pump YdhE/NorM, homolog, Cassette chromosome recombinase B, Antiadhesin Pls, binding to squamous nasal epithelial cells, Arsenical resistance operon repressor, PlcR-regulated protein PRP2, Na+/H+ antiporter, SceD-like transglycosylase, biomarker for vancomycin-intermediate strains. The isolate T28b had 23 singletons with Bacitracin transport ATP-binding protein bcrA, Universal stress protein family and, Fosfomycin resistance protein FosB among the singletons. In isolate T27 and T28 singletons were mainly of unknown function. In all isolates from South Africa, tetM gene was detected (part of the accessory genome), but it was not detected in any of the isolates from Nigeria (see Table 3). In addition, S. cohnni isolates from Nigeria had at least one gene for cadmium resistance in their accessory genome or singletons, these genes were not detected in isolates from South Africa. Isolates L6 and T4 were identified as S. haemolyticus, though they were also from distinct geographic origins, we did not do the pan-genome analysis because their genomes were estimated to be incomplete.
Antibiotic resistant genes identified from the isolates.
| Isolate | RGI Criteria | ARO term | AMR Gene Family | Drug Class | Resistance Mechanism | Matching Region | Ref Seq Length |
|---|---|---|---|---|---|---|---|
| T20 | perfect | mecl | methicilin resistant PBP2 | penam | antibiotic target replacement | 100 | 100 |
| Strict | tetM | tetracylin-resistant ribosomal protection protein | tetracyclin antibiotic | antibiotic target protection | 97.65 | 100 | |
| Strict | mecA | methicilin resistant PBP2 | penam | antibiotic target replacement | 99.85 | 100 | |
| Strict | mphC | macrolide phosphotransferase (MPH) | macrolide antibiotic | antibiotic inactivation | 90.64 | 100 | |
| Strict | Erm(43) | Erm 23S ribosomal RNA methyltransferase | macrolide antibiotic, lincosamide antibiotic, streptogramin antibiotic | antibiotic target alteration | 98.77 | 100 | |
| Srict | ErmA | Erm 23S ribosomal RNA methyltransferase | macrolide antibiotic, lincosamide antibiotic, streptogramin antibiotic | antibiotic target alteration | 99.59 | 100 | |
| Strict | ErmB | Erm 23S ribosomal RNA methyltransferase | macrolide antibioyic, lincosamide antibiotic, streptogramin antibiotic | antibiotic target alteration | 98.78 | 98.79 | |
| L4 | Perfect | dfrG | trimethoprim resistant dihydrofolate reductase dfr | diaminopyrimidine antibiotic | antibiotic target replacement | 100 | 100 |
| Strict | PC1 beta- lactamase (blaZ) | blaZ beta lactamase | penam | antibiotic inactivation | 95.37 | 100 | |
| d13b | Strict | FusF | fusidic acid inactivation enzyme | fusidic acid | antibiotic inactivation | 99.07 | 100 |
| T28b | Perfect | FosB1 | fosfofomycin thiol transferase | fosfomycin | antibiotic inactivation | 100 | 100 |
| Strict | FusF | fusidic acid inactivation enzyme | fusidic acid | antibiotic inactivation | 99.53 | 100 | |
| Strict | FosD | fosfofomycin thiol transferase | fosfomycin | antibiotic inactivation | 96.4 | 100 | |
| T27 | Srict | FusF | fusidic acid inactivation enzyme | fusidic acid | antibiotic inactivation | 99.53 | 100 |
| Strict | tetM | tetracylin-resistant ribosomal protection protein | tetracyclin antibiotic | antibiotic target protection | 97.81 | 100 | |
| T6 | Strict | PC1 beta-lactamase (blaZ) | blaZ beta lactamase | penam | antibiotic inactivation | 96.09 | 100 |
| T28 | Strict | FusF | fusidic acid inactivation enzyme | fusidic acid | antibiotic inactivation | 99.53 | 100 |
| Strict | FosD | fosfofomycin thiol transferase | fosfomycin | antibiotic inactivation | 96.4 | 100 | |
| Strict | tetM | tetracylin-resistant ribosomal protection protein | tetracyclin antibiotic | antibiotic target protection | 97.81 | 100 | |
| C33 | n/a | n/a | n/a | n/a | n/a | n/a | n/a |
Mobile Genetic elements
Two intact prophage regions were identified by PHASTER analysis for two (L4 and C33) of the eight isolates. L4 and C33 prophage regions had a total length of 40.1kb and 58.5kb, encoding 59 and 75 proteins, respectively. For L4 the most common phage name was PHAGE_Staphy_StB12_NC_020490, whereas for C33 it was PHAGE_Staphy_phiRS7_NC_022914. Of the proteins encoded by L4 prophage region, peptidoglycan hydrolase, Erf-like recombinase, putative cro/cl-like repressor, LexA repressor, pyrophosphatase, pathogenicity island protein, and mobile-element-associated regulatory protein were of interest. In contrast, C33 encoded holin, dihydrofolate reductase type 1, protein phosphatase, and serine/threonine kinase.
PlasmidFinder detected a total of nine plasmids distributed among five of the eight isolates (Table 1). D13b plasmid was identical to S. epidermidis plasmid SAP016A (GQ900381), while that of T6 was identical to S. epidermidis plasmid SAP105A (GQ900452). Isolate T27 and T28 had two similar plasmids which were identical to S. saprophyticus. subsp saprophyticus ATCC 15305 plasmid pSSP1 (AP008935) and Staphylococcus sp. 693-2 plasmid pLEW6932 (NC_009130.1). The isolate T28b had three plasmids identical to Staphylococcus aureus subsp. aureus ST398 pS0385-3 plasmid, isolate SO385 (AM990995.1), Staphylococcus saprophyticus pSES22 plasmid, isolate 44 (AM159501.1) and Staphylococcus saprophyticus subsp. saprophyticus ATCC 15305 plasmid pSSP1 (AP008935.1). We were mainly interested in virulence and antibiotic resistance genes carried on the plasmids or mobile elements since they can be easily transferred to other microbes in the environment. Only T28b plasmid carried a gene of interest (FosB), which was on the plasmid identical to Staphylococcus saprophyticus subsp. saprophyticus ATCC 15305 plasmid pSSP1 (AP008935.1). Though no plasmid was detected for T20, the genome had an insertion sequence (IS256-like element ISLgar5 family transposase) which carried the mecA gene.
Resistome and Virulome
A number of antibiotic resistance-associated genes were identified by using the Comprehensive Antibiotic Resistance Database (CARD). Table 3 shows the predicted antibiotic-resistance genes, the mechanism of resistance as well as the drug class for each isolate. All of the isolates except for C33 had at least one hit of the antibiotic resistant genes. All the resistome and virulome genes were located on the chromosomes except for FosB in isolate T28b which was located on the plasmid as well as mecA in isolate T20 which was located on an insertion sequence.
The Virulence Factors of Pathogenic Bacteria Database (VFDB) was used to determine the potential virulome of the isolates. All the isolates had at least one virulence factor (Table 4a and 4b). Interestingly, the analysis showed that some of the virulence factors were acquired from non-Staphylococcus spp (Table 4b) suggesting the possibility of these factors being acquired through horizontal gene transfer. The factors covered a broad range of categories which include adherences, enzymes, immune invasion, toxin, surface protein anchoring, anti-phagocytosis, and phagosome arrest among other categories.
Distribution of virulence factors which are largely confined to the Staphylococcus genus.
| L4 | D13b | T28 | T28b | C33 | T20 | T6 | T27 | S. aureus (subsp aureus) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Adherence | ||||||||||
| Autolysin | (atl) | + | + | + | + | + | - | + | + | SACOL1062 |
| Clumping factor B | (clfB) | + | - | - | - | - | + | - | - | SACOL2652 |
| Elastin binding protein | (ebp) | + | + | + | + | - | - | + | + | SACOL1522 |
| Enzymes | ||||||||||
| Lipase | (lip) | + | + | + | + | - | - | - | + | SACOL2694 |
| Serine V8 protease | (sspA) | - | + | + | + | + | + | + | + | SACOL1057 |
| Thermonuclease | (nuc) | + | + | + | + | + | - | + | + | |
| Immune invasion | SACOL0860 | |||||||||
| Capsule | +(2) | +(5) | +(6) | +(6) | - | +(3) | +(2) | +(6) | SACOLO136 -141 | |
Distribution of virulence factors likely to have been acquired from non-Staphylococcus genomes.
| L4 | D13b | T28 | T28b | C33 | T20 | T6 | T27 | Organism | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Imune invasion | ||||||||||
| Capsule | - | - | + | + | - | - | + | Acinetobacter | ||
| Polysacharide capsule | - | - | - | - | - | + | - | Bacilus | ||
| Toxin | ||||||||||
| Cytolisin | (cyIR2) | - | - | + | + | + | - | + (4) | Enterococcus | |
| Nutrional factor | ||||||||||
| Allantoin utilisation | - | + (4) | + (4) | + (4) | + (4) | - | + | Klebsiela | ||
| Serum resistance and immune invasion | ||||||||||
| LPS | (wbtE) | - | + | - | - | - | - | - | (Francisella) | |
| LPS | (wbtP) | - | - | - | - | - | + | - | (Francisella) | |
| Antiphagocytocysis | ||||||||||
| Capsule | (uge) | - | - | + | + | - | - | + | Klebsiela | |
| Iron uptake | ||||||||||
| Periplasmic binding protein-dependent ABC transport systems | (vctC) | - | - | - | - | - | + | - | Vibrio | |
| Phagosome arresting | ||||||||||
| Nucleoside diphosphate kinase | (ndk) | - | - | - | - | - | + | - | Mycobacterium | |
| Regulation | ||||||||||
| LisR/LisK | (lisR) | - | - | - | - | - | + | - | Listeria | |
| Surface protein anchoring | ||||||||||
| Lipoprotein diacylglyceryl transferase | (lgt) | - | - | - | - | - | + | - | Listeria | |
Discussion
In the current study, whole-genome sequencing of eight coagulase-negative staphylococci (CoNS) isolates was performed to characterise the antibiotic resistance and virulence factors associated with the isolates and the potential dangers they may pose. The isolates were from two distinct geographical locations (Supplementary Table 1) in which cadmium pollution and abuse of antibiotics were reported, thus we hypothesised that these anthropogenic activities are likely to influence the genomic features of the isolates. The pan-genomic approach was applied to highlight the variation of the isolates. Five of the eight isolates (T27, T28, T28b, C33, and D13b) were identified as S. cohnni (Fig. 1 and Table 2). Of these isolates, C33, D13b, and T28b were isolates from Nigeria, the remaining (T27, T28) were from South Africa. Isolates from South Africa (T20, T27, and T28) carried the tetracycline-resistant ribosomal protection protein (tetM) which was not detected from any of the samples from Nigeria. Phylogeographic structure, regional or continental endemism have been demonstrated in a number of bacterial species [32, 33]. This correlates with the accessory genomes which are regarded as essential for survival under various selective pressures. In the current study, the tetM gene was part of the accessory genome, large fraction of the accessory genome are mobile genetic elements [34]. These accessory genomes place the host cell in an advantageous position to be viable under specific conditions [35, 36]. The elevated levels of tetracycline and other antibiotics in the wastewater treatment plants in the North West province, South Africa [12], could have exerted a selective pressure, which resulted in the acquisition of the tetM accessory genome probably through horizontal gene transfer. This presumption is also supported by the presence of fewer barriers to horizontal gene transfer among the genomes.
Environmentally driven heterogeneity among bacteria represents the physiological response to stress [37], and survival strategies developed over evolutionary time [38]. All S. cohnii isolates from Nigeria possessed at least one gene for cadmium tolerance or resistance in their accessory genomes or singletons. Cadmium is one of the major driving forces for bacterial selection and evolution in the area where the isolates were collected (Nigeria). A number of studies have reported extreme soil and surface water contamination with heavy metals (including cadmium) in and around Ile-Ife, Nigeria [13, 14, 15, 16]. The genomic heterogeneity among the samples from South Africa and Nigeria, clearly highlights how anthropogenic activity can shape the genomes of microbes in our surroundings.
Isolates used in this study possess an open pan-genome, increasing the number of species in the analysis resulted in an increase in the number of distinct genes (Fig. 3). Most pathogenic bacteria possess an open pan-genome, with the accessory genomes mainly consisting of virulent determinants and antibiotic-resistant genes on mobile genetic elements (MGEs) which would have been acquired through horizontal gene transfer. MGEs normally confers a number of adaptive advantages under a particular environment and support the virulence of organisms [39]. In CoNS, MGEs have been shown to bear S. aureus pore-forming toxins and super-antigen enterotoxin coding sequences [40], which have been shown to modulate the virulence of CoNS bacteria [41]. Isolates C33 and D13b had a significantly higher proportion of singletons compared to isolates T27, T28, and T28b. This was expected since the isolates were of clinical origin, and the singletons were presumed to be factors important for infection and hospital adaptation. True to this, the singletons consisted of virulence factors such as integrase, super-antigen encoding pathogenicity islands (SaPI), antimicrobial resistance (Undecaprenyl-diphosphatase (EC 3.6.1.27), PBP2a, MecA, MecR1, Blal, aadA), adaptations to stress (cadmium resistance protein, EfeB, abortive phage resistant protein, putative cytoplasmic protein).
Phylogenetic pan-genome accumulation of the S. cohnni isolates used in this study and the reference genome S. cohnni subsp cohnii (GCF_000972575.1). Changes in the pan-genome are highlighted per each node.
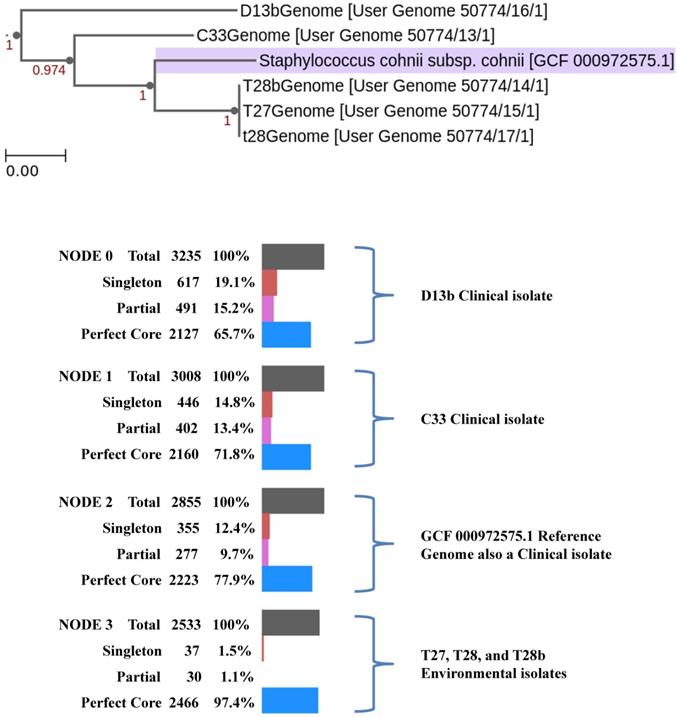
Only one (T20) out of the eight isolates used in this study possessed the CRISPR/Cas system which is a strong barrier to foreign DNA uptake, in particular plasmid DNA [42, 43, 44]. Consistent with this, T20 had no detected phages or plasmids (Table 1) however, T20 consisted of SaPIs, leading to the hypothesis that this pathogenic island might have been acquired before acquisition of the CRISPR/Cas region. CoNS have been shown to harbour fewer CRISPR/Cas systems compared to other bacteria [45]. This has been linked to a potential role as a reservoir for antibiotic-resistant genes for S. aureus [46] since they can easily acquire these genes from the environment and transfer them to S. aureus through horizontal gene transfer. Intact prophages were only detected in isolates (L4 and C33). Prophages are known to increase the virulence potential of pathogenic strains [47, 48], and there is a positive correlation between the phage-related DNA content of a given Enterobacteria and its pathogenicity [49]. In this study, phage-related DNA constituted a significant portion of the singletons among isolates, this was expected particularly in isolates of clinical origin.
CoNS are important reservoirs for antibiotic-resistant genes, including ß-lactamase genes. These are mainly carried on mobile genetic elements and can easily be transferred to highly virulent species such as S. aureus [50]. The continuous spread of Methicillin-Resistant Staphylococcus aureus (MRSA) and Methicillin-Resistant Coagulase Negative Staphylococci (MR-CoNS) strains in clinical and non-clinical environments remain a serious public health concern. This is mainly due to the difficulties associated with the treatment of infections from such organisms as they become resistant to methicillin which is probably considered the prototype of anti-staphylococci penicillin [51]. Methicillin resistance is characterised by an altered penicillin-binding protein (PBP2a) which has a reduced affinity for methicillin rendering the drug ineffective [4, 52, 53]. PBP2a is encoded by mecA which is normally acquired through horizontal gene transfer of a mobile genetic element staphylococci cassette chromosome mec (SCCmec) [54]. In the present study, mecA was detected only in isolate T20 designated as S. lentus (Table 3). The isolate also had multiple other antibiotic resistance genes which included tetracycline-resistant ribosomal protection protein (tetM), macrolide phosphotransferase (mphC) Erm 23S ribosomal RNA methyltransferase(s) (Erm43/A/B). This augments the ever-increasing evidence of wastewater treatment plants being a source of multiple antibiotic resistance genes. A diverse group of antibiotics targets the ribosome as the major site of action in the bacterial cell [55], thus altering and protecting these target sites is vital for bacterial survival in the face of antibiotics. Methylation of rRNA provides acquired antibiotic resistance [56], thus the Erm group provides a crucial role in acquired resistance. Tetracycline, a broad-spectrum antibiotic agent binds to elongating ribosome and inhibit delivery of the ternary complex to the A-site [57] thus inhibiting bacterial translation and slowly killing it. Continuous use and abuse of tetracycline resulted in bacteria acquiring resistance determinants to this group of antibiotics [58]. The resistance determinants mainly include ribosome protection proteins with the most prevalent being tetM and tetO [59, 60]. The tetM was also detected in S. cohnii isolates (T27 and T28; Table 3). Also of interest was the detection of genes associated with ß-lactamase activities. The isolates identified as S. haemolyticus carried the PC1 ß-lactamase (blaZ) gene (Table 3). Table 3 gives an overview of the detected genes providing resistance to various antibiotics. This table also provides information on drug class and resistance mechanism.
Eighty percent of the S. cohnii isolates contained the FusF gene. These were from both Nigeria and South Africa. Resistance to fusidic acid is selected for by topical antibiotic use. An increase in the emergence of fusidic acid resistance among clinical isolates which were attributed to the widespread and inappropriate use of fusidic acid cream for chronic skin conditions has been recently reported [61]. Normally, resistance is conferred by genes encoding FusB-family proteins through the protection of the drug target site [62]. However, Chen et al., [63] identified a novel fusidic acid resistance determinant, FusF, in fusidic acid resistant S. cohnii. The gene is thought to be a natural part of the S. cohnii genome. From the study, only S. cohnii carried this gene out of a number of CoNS species, and this was similar to the present study.
Conclusion
In conclusion, anthropogenic activities seemed to impact the accessory genome, contributing to the variation of isolates within the same species. CoNS isolates used in this study possessed an open pan-genome with few barriers for horizontal gene transfers. Thus the isolates could easily acquire foreign DNA, which helps to adapt to the selective pressure, this explains the observed environmentally driven heterogeneity. Pan-genome analysis proved to be a powerful tool for comparative genomics as it helped to highlight the environmentally driven heterogeneity of the isolates. Moreover, the clinical isolates could be clearly distinguished from the environmental isolates based on the composition of the singletons. More interestingly, was the presence of virulence determinants and multiple antibiotic resistance genes from environmental isolates, demonstrating the importance of wastewater treatments as a source of antibiotic resistant genes. This study adds to the baseline for antibiotic resistance surveillance and comparative genomics of potentially pathogenic CoNS.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding
The authors acknowledge with thanks the financial support from the North-West University, South Africa, the research support in part by the National Research Foundation (NRF) of South Africa (Grant No. 113824), and the Water Research Commission (WRC), South Africa (project K5/2347/3). Views expressed are those of the authors and not of the funders.
Availability of data and materials
The raw sequencing data generated by Illumina MiSeq 2000 were deposited to the National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under BioProject ID PRJNA607879 with Accession No (SAMN14149990 - SAMN14149997).
Ethical statement
This study was approved by the ethics committee of the hospital used in Ile-Ife, Nigeria, under protocol number: SERC-2016-001-NWU-SA and Health Research Ethics Committee (HREC) of the Faculty of Health Sciences, North-West University, Potchefstroom, South Africa, under ethics number: NWU-00122-17-S1. All clinical staphylococcal isolates evaluated in this study were obtained from the Microbiology Laboratory of the Medical Institution, Ile-Ife, Nigeria, and were anonymized.
Health and safety
The authors declare that all mandatory laboratory health and safety procedures and protocols were complied with while conducting the experimental work reported in this study.
Author Contributions
CCB and TEM conceptualized the study, CCB supervised and secured the funds for the study. AOO collected the samples and performed the experiments. TEM performed bioinformatics analysis and drafted the manuscript. All authors read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Banerjee SN, Emori TG, Culver DH, Gaynes RP, Jarvis WR, Horan T. et al. Secular trends in nosocomial primary bloodstream infections in the United States, 1980-1989. National Nosocomial Infections Surveillance System. Am J Med. 1991;91:86S-89S
2. Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;38:309-317
3. Otto M. Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu Rev Med. 2013;64:175-188
4. Turner NA, Sharma-Kuinkel BK, Maskarinec SA, Eichenberger EM, Shah PP, Carugati M. et al. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat Rev Microbiol. 2019;17:203
5. Lee BY, Singh A, David MZ, Bartsch SM, Slayton RB. et al. The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). Clin Microbiol Infect. 2013;19:528-536
6. Gajdács M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics (Basel). 2019;8:52
7. Wang SM, Liu CC, Tseng HW, Yang YJ, Lin CH, Huang AH. et al. Staphylococcus capitis bacteremia of very low birth weight premature infants at neonatal intensive care units: clinical significance and antimicrobial susceptibility. J Microbiol Immunol Infect. 1999;32:26-32
8. Brito DV, von Dolinger EJ, Abdallah VO, Darini AL, Gontijo Filho PP. Two outbreaks of mixed etiology associated with central venous catheters inserted by phlebotomy in critical neonates. Braz J Infect Dis. 2009;13:177-182
9. Mazzariol A, Lo CG, Kocsis E, Maccacaro L, Fontana R, Cornaglia G. Outbreak of linezolid-resistant Staphylococcus haemolyticus in an Italian intensive care unit. Eur J Clin Microbiol Infect Dis. 2012;31:523-527
10. Miragaia M, Couto I, de Lencastre H. Genetic diversity among methicillin-resistant Staphylococcus epidermidis (MRSE). Microb Drug Resist. 2005;11:83-93
11. Zong Z, Peng C, Lü X. Diversity of SCCmec elements in methicillin-resistant coagulase-negative staphylococci clinical isolates. PLoS One. 2011;6:e20191
12. Kanama KM, Daso AP, Mpenyana-Monyatsi L, Coetzee MAA. Assessment of pharmaceuticals, personal care products, and hormones in wastewater treatment plants receiving inflows from health facilities in North-West province, South Africa. J Toxicol. 2018;2018:3751930
13. Fatoki OS. Levels of dissolved zinc and cadmium in some surface waters of western Nigeria. Environ Int. 1993;19:285-289
14. Oladipo OG, Olayinka A, Awotoye OO. Ecological impact of mining on soils of Southwestern Nigeria. Environ Exper Biol. 2014;12:179-186
15. Eludoyin AO, Ojo AT, Ojo TO, Awotoye OO. Effects of artisanal gold mining activities on soil properties in a part of southwestern Nigeria. Cogent Environ Sci. 2016;3:1305650
16. Joe-Ukairo A, Oni AG. Geophysical and hydro-chemical investigation of Oke Asunle dumpsite in Ile-Ife, southwestern Nigeria for subsoil and surface water pollution. J Health Pollut. 2018;20:181209
17. Oladipo AO, Oladipo OG, Bezuidenhout CC. Multi-drug resistance traits of methicillin-resistant Staphylococcus aureus and other Staphylococcal species from clinical and environmental sources. J Water Health. 2019;17:930-943
18. Isenberg HD. Identification of commonly-isolated aerobic Gram-positive bacteria. In Clinical Microbiology Procedures Handbook. 1992 pp. 1.20.1-1.20.12. Edited by H. D. Isenberg. Washington, DC: American Society for Microbiology
19. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-2120
20. Arkin AP, Cottingham W, Henry CS, Harris NL, Stevens RL, Maslov S. et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat Biotechnol. 2018;36:566-569
21. Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A. et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Compt Biol. 2013;20:714-737
22. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043-1055
23. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068-2069
24. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA. et al. 2008. The RAST Server: Rapid annotations using subsystem technology. BMC Genomics. 2008;9:75
25. Price MN, Dehal PS, Arkin AP. FastTree 2 - Approximately Maximum-Likelihood Trees for Large Alignments. PLoS One. 2010;5:e9490
26. Jain C, Rodriguez-R LM, Phillipy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2017;9:5114
27. Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;8:W16-21
28. Carattoli A, Zankari E, Garcia-Fernandez A, Larsen MV, Lund O, Villa L. et al. In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob Angents Chemother. 2014;58:3895-3903
29. Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y. et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2016;1:D3253-28
30. Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A. et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acid Res. 2020;48:D517-D525
31. Couvin D, Bernheim A, Toffanonioche C, Touchon M, Michalik J, Néron B. et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46:W246-51
32. Cho J, Tiedje JM. 2000. Biogeography and degree of endemicity of fluorescent Pseudomonas strains in soil. Appl Environ Microbiol. 2000;66:5448-5456
33. Zwirglmaier K, Jardillier L, Ostrowski M, Mazard S, Garczareck L, Not F. et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 2008;10:147-161
34. Frost LS, Leplae R, Summers OA, Tossaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3:722-732
35. Mira M, Martin-Cuadrado AB, Auria GD, Rodriguez-Valera F. 2010. The bacterial pan-genome: a new paradigm in microbiology. Int Microbiol. 2010;13:45-57
36. Read TD, Ussery DW. Opening the pan-genomics box. Curr Opin Microbiol. 2006;9:496-498
37. Ryall B, Eydallin G, Ferenci T. Culture history and population heterogeneity as determinants of bacterial adaptation: the adaptomics of a single environmental transition. Microbiol Mol Biol Rev. 2012;76:597-625
38. Booth IR. Stress and the single cell: intrapopulation diversity is a mechanism to ensure survival upon exposure to stress. Int J Food Microbiol. 2002;78:19-30
39. Bosi E, Monk JM, Aziz RK, Fondi M, Nizet V, Palsson BO. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. PNAS. 2016;113:E3801-E3809
40. Sato'o Y, Omoe K, Ono HK, Nakane A, Hu D-L. A novel comprehensive analysis method for Staphylococcus aureus pathogenicity islands. Microbiol Immunol. 2013;57:91-99
41. McCarthy AJ, Loeffler A, Witney AA, Gould KA, Lloyd DH, Lindsay JA. Extensive horizontal gene transfer during Staphylococcus aureus co-colonization in vivo. Genome Biol Evol. 2014;6:2697-708
42. Cao L, Gao C-H, Zhu J, Zhao L, Wu Q, Li M. et al. Identification and functional study of type III-A CRISPR-Cas systems in clinical isolates of Staphylococcus aureus. Int J Med Microbiol. 2016;306:686-96
43. Li Q, Xie X, Yin K, Tang Y, Zhou X, Chen Y. et al. Characterization of CRISPR-Cas system in clinical Staphylococcus epidermidis strains revealed its potential association with bacterial infection sites. Microbiol Res. 2016;193:103-10
44. Louwen R, Staals RHJ, Endtz HP, van Baarlen P, Van der Oost J. The role of CRISPR-Cas Systems in Virulence of pathogenic Bacteria. Microbiol Mol Biol Rev. 2014;78:74-88
45. Rossi CC, Souza-Silva T, Araújo-Alves AV, Giambiagi-deMarval M. CRISPR-Cas Systems Features and the Gene-Reservoir Role of Coagulase-Negative Staphylococci. Front Microbiol. 2017;8:1545
46. Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67-78
47. Boyd EF. Bacteriophage-encoded bacterial virulence factors and phage-pathogenicity islands interactions. Adv Vir Res. 2012;82:91-118
48. Varani AM, Monteiro-Vitorello CB, Nakaya HI, Van Sluys MA. The role of prophage in plant pathogenic bacteria. Annu Rev Phytopathol. 2013;51:429-451
49. Bobay LM, Rocha EPC, Touchon M. The adaptation of temperate bacteriophages to their host genomes. Mol Biol Evol. 2013;4:737-751
50. Becker K, Heilmann C, Peters G. Coagulase-negative staphylococci. Clin Microbiol Rev. 2014;27:870-926
51. Lobanovska M, Pilla G. 2017. Penicillin's Discovery and Antibiotic Resistance: Lessons for the Future? Yale J Biol Med. 2017;90:135-145
52. Eady EA, Cove JH. Staphylococcal resistance revisited: Community-acquired methicillin resistant Staphylococcus aureus-an emerging problem for the management of skin and soft tissue infections. Curr Opin Infect Dis. 2003;16:103-124
53. Sabath LD, Finland M. Inactivation of methicillin, oxacillin and ancillin by Staphylococcus aureus. Proc Soc Exp Biol Med. 1962;111:547-550
54. Katayama Y, Ito T, Hiramatsu K. A new class of genetic element, staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 2000;44:1549-1555
55. Poehlsgaard J, Douthwaite S. 2005. The bacterial ribosome as a target for antibiotics. Nat Rev Microbiol. 2005;3:870-881
56. Douthwaite S, Crain PF, Liu M, Poehlsgaard J. The tylosin-resistance methyltransferase RlmAII (TlrB) modifies the N-1 position of 23S rRNA nucleotide G748. J Mol Biol. 2004;337:1073-1077
57. Wilson DN. The A-Z of bacterial translation inhibitors. Crit Rev Biochem Mol Biol. 2009;44:393-433
58. Roberts MC. 2005. Update on acquired tetracycline resistance genes. FEMS Microbiol Lett. 2005;245:195-203
59. Chopra I, Roberts M. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev. 2001;65:232-260
60. Connell SR, Tracz DM, Nierhaus KH, Taylor DE. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob Agents Chemother. 2003;47:3675-3681
61. Dobie D, Gray J. Fusidic acid resistance in staphylococcus aureus. Arch Dis Child. 2004;89:74-77
62. O'Neill AJ, Chopra I. Molecular basis of fusB mediated resistance to fusidic acid in Staphylococcus aureus. Mol Microbiol. 2005;59:664-676
63. Chen H, Hung W, Lin Y, Tsai J, Chiu H, Hsueh P. et al. A novel fusidic acid resistance determinant, fusF, in Staphylococcus cohnii. J Antimicrob Chemother. 2015;70:416-419
Author contact
![]() Corresponding author: Tawanda Elias Maguvu (temaguvucom)
Corresponding author: Tawanda Elias Maguvu (temaguvucom)