ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2018; 6:63-73. doi:10.7150/jgen.24368 This volume Cite
Research Paper
Isolation and Identification of an Anthracimycin Analogue from Nocardiopsis kunsanensis, a Halophile from a Saltern, by Genomic Mining Strategy
Fernanda L. Sirota1,# ![]() , Falicia Goh1,#, Kia-Ngee Low1,#, Lay-Kien Yang1, Sharon C. Crasta1, Birgit Eisenhaber1, Frank Eisenhaber1,2, Yoganathan Kanagasundaram1
, Falicia Goh1,#, Kia-Ngee Low1,#, Lay-Kien Yang1, Sharon C. Crasta1, Birgit Eisenhaber1, Frank Eisenhaber1,2, Yoganathan Kanagasundaram1 ![]() , Siew Bee Ng1
, Siew Bee Ng1 ![]()
1. Bioinformatics Institute, Agency for Science, Technology and Research (A*STAR), 30 Biopolis Street, #07-01 Matrix, Singapore 138671, Republic of Singapore
2. School of Computer Engineering, Nanyang Technological University (NTU), 50 Nanyang Drive, Singapore 637553, Republic of Singapore
# Authors contributed equally to this work
Received 2017-12-14; Accepted 2018-4-25; Published 2018-5-21
Abstract

Modern medicine is unthinkable without antibiotics; yet, growing issues with microbial drug resistance require intensified search for new active compounds. Natural products generated by Actinobacteria have been a rich source of candidate antibiotics, for example anthracimycin that, so far, is only known to be produced by Streptomyces species. Based on sequence similarity with the respective biosynthetic cluster, we sifted through available microbial genome data with the goal to find alternative anthracimycin-producing organisms. In this work, we report about the prediction and experimental verification of the production of anthracimycin derivatives by Nocardiopsis kunsanensis, a non-Streptomyces actinobacterial microorganism. We discovered N. kunsanensis to predominantly produce a new anthracimycin derivative with methyl group at C-8 and none at C-2, labeled anthracimycin BII-2619, besides a minor amount of anthracimycin. It displays activity against Gram-positive bacteria with similar low level of mammalian cytotoxicity as that of anthracimycin.
Keywords: Anthracimycin, Nocardiopsis, gene cluster, sequence analysis, biosynthesis
Introduction
The biosynthetic gene cluster of the macrolide anthracimycin (atc) has been identified in less than a handful of bacterial strains belonging to Streptomyces sp. It was not long ago that it was first reported for the marine strain Streptomyces sp. T676 isolated off St. John's Island in Singapore and described as a type I trans-AT PKS. [1]. In contrast to cis-AT PKSs, trans-AT PKS enzymes show an amount of architectural diversity that makes the cis-AT co-linearity rule not directly applicable. For instance, they can have a cross-module domain mode of action, unique domains, split modules between two proteins, despite their trans-acting functionality and other additional unusual features [2]. In this regard, evolution by horizontal gene transfer and extensive recombination of PKS gene fragments seem to be a preferred mode of action by trans-AT PKSs [2,3].
Historically, the atc compound (1 in Figure 1) was discovered by MerLion Pharmaceuticals Pte Ltd as a result of a high-throughput screening for new antibacterials (unpublished data) in 2001. However, it was only in 2013 that its chemical structure was reported, when the compound was isolated from Streptomyces sp. CNH365 [4], a marine strain found in sediments near-shore off Santa Barbara, CA. In 2014, the year before the atc cluster was reported, the genome of Streptomyces sp. NRRL F-5065 became available (accession no. JOHV00000000) [5]. In 2015, the draft genome of another anthracimycin producing strain, Streptomyces sp. TP-A0875, isolated from a compost sample in Ishikawa, Japan, was published [6,7]. In 2008, the structure of chlorotonil A (3 in Figure 1), a compound isolated from the myxobacterium Sorangium cellulosum has been reported [8]. Both anthracimycin and chlorotonil A are tricyclic metabolites with a similar carbon skeleton; chlorotonil A seems to be almost the optical isomer of anthracimycin [6]. Its difference from anthracimycin includes its unique gem-dichloro-1,3-dione moiety and a methyl group in position C-8. The biosynthetic gene cluster for chlorotonil A has been published at the same time as the one for anthracimycin and was shown to also produce new chlorotonil congeners. Its cluster organization, as well as its proposed pathway, showed some striking differences to that of anthracimycin [9].
Chemical structures of compounds. Anthracimycin (1) produced by Streptomyces sp. T676, anthracimycin BII-2619 (2) produced by Nocardiopsis kunsanensis DSM 44524 and chlorotonil A (3) produced by Sorangium cellulosum So ce1525.
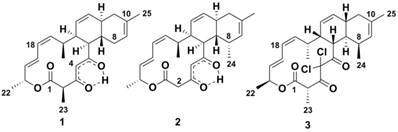
The recent increase in attention to anthracimycin is due to its strong activity against Gram-positive bacteria, including some drug-resistant strains such as Bacillus anthracis, methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE), its rapid bacterial killing kinetics and in vivo efficacy in the murine peritonitis model [4,10]. Chlorotonil A has most notably shown potent bioactivity against Plasmodium falciparum, one of the malaria pathogens [11]. Thus, the search for different organisms producing the same compound or its analogues is of great importance for future advances, as different organisms might lead to greater compound production yields or generate other biologically effective derivatives.
We searched for microbial genomes that contain sequence segments similar to the known atc biosynthetic gene cluster in Streptomyces. In this work, we report the prediction of a potential anthracimycin derivative-producing gene cluster in Nocardiopsis kunsanensis, an actinobacterial microorganism belonging to Streptosporangiales, a different order compared to the reported anthracimycin-producing Streptomyces. Most interestingly, experimental verification of this finding revealed that N. kunsanensis predominantly synthesized a new anthracimycin derivative and only a very small amount of anthracimycin. After the elucidation of its structure, we named this new analogue compound anthracimycin BII-2619 (see 2, Figure 1). We also compared its biological activity with anthracimycin.
Methods
Bioinformatics search methods: prediction of the anthracimycin (atc) gene cluster in Streptosporangiales
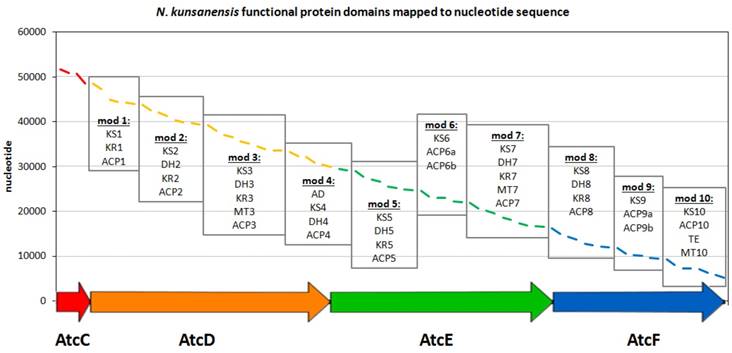
In order to identify other potential anthracimycin producing microorganisms, we first focused on the sequences of the functional domains as defined for Streptomyces sp. T676 in the public database (accession no. LN871452). For this entry, there are nine polyketide synthase (PKS) gene products (atcA, atcB, atcC, atcD, atcE, atcF, atcG, atcH, atcI), where four of them (atcC-atcF) have been proposed to be functionally involved in the generation of anthracimycin [1]. These four products were further split into modules according to the definition of functional modules commonly present in biosynthetic megaenzymes, where independently folding protein domains are joined together via so-called 'linker' regions [12]. The boundaries of these domains were annotated in the entry as misc_feature. For the atcD gene product, an extra feature was annotated as “repeated sequence; additional in PacBio sequence”. This region of 62 residues is rich in low complexity, as seen in the ANNOTATOR platform [13] using a consensus disorder prediction [14], and so was the only region not included in this analysis. All other 41 domains were blasted and psi-blasted [15] against nr (Dec 2015) [16], so as to identify species that shared the highest number of domains significantly similar to the ones in the atc cluster. 1292 and 2000 organisms were found by blast and psi-blast (E-value cut off 0.001), respectively, to produce at least 1 significant domain hit. In this work, we limited ourselves to display the top strains that displayed 39 or more hits to all domains present in the atc cluster (Supplementary Table S1). In order to identify if the protein domains found in N. kunsanensis would have the architecture of a biosynthetic gene cluster, tblastn was used with the annotated T676 protein domains as queries, to map them in the N. kunsanensis nucleotide sequence. All the protein domains had a significant hit (E-value cut off 0.001) and could be mapped to N. kunsanensis DSM 44524 contig 32 (accession no. NZ_ANAY01000032) (Figure 2). All the regions plotted in Figure 2 refer to the first tblastn hit result, with the exception of domains DH5 (in AtcE) and KR8 (in AtcF) that were the 2nd and 3rd best hits, respectively.
Organization of the N. kunsanensis atc biosynthetic cluster (AtcC-AtcF). The functional protein domains are mapped to the nucleotide (complementary) sequence of N. kunsanensis DSM 44524 contig 32 (NZ_ANAY01000032) and are colored in red, orange, green and blue regarding the proteins they belong to. The PKS modules are indicated by boxes, and the functional domains are listed accordingly: ACP, acyl-carrier protein; AD, alcohol dehydrogenase; DH, dehydratase; KR, β-ketoreductase; KS, β-ketoacyl synthase; MT, methyltransferase; TE, thioesterase. AtcC harbours the two acyltransferase domains, AT1, and AT2-ER (ER, enoylreductase).
Sequence alignment and phylogenetic analysis
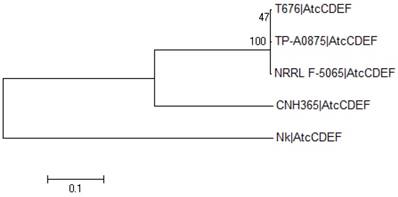
Each one of the four functional proteins in the cluster (atcC, atcD, atcE and atcF) had their amino acid sequences aligned among the five strains with the MAFFT E-INS-i algorithm [17]. Individual phylogenetic trees were calculated with MEGA6 [18] for each of the four aligned proteins. These included Neighbor-Joining and Maximum Likelihood with and without bootstrap methods (data not shown). Given that there was no observed difference among the generated trees for each of these four proteins, regardless of the method, these aligned sequences were intentionally merged so as to produce a general picture of the evolutionary relationship of these four functional proteins among the five strains that reflects their individual protein trees (Figure 3).
Phylogenetic analysis of atc cluster sequences from different organisms. The evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model. The tree with the highest log likelihood (-87859.2839) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using a JTT model. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 1.6861)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 20.9551% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 5 amino acid sequences of merged proteins AtcC-AtcF, labelled as AtcCDEF. All positions containing gaps and missing data were eliminated. There was a total of 15045 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 [18].
Bacterial strain acquisition and fermentation conditions
Nocardiopsis kunsanensis DSM 44524 was purchased from Leibniz-Institut DSMZ - Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ) [19]. Nocardiopsis kunsanensis DSM 44524 was grown on Complex Medium (CM) agar for 7 days at 28°C and was used to prepare 50 mL seed culture in CM (7.5 g casamino acids, 10 g yeast extract, 3 g sodium citrate, 10 g magnesium sulfate, 2 g potassium chloride, 1 mL 4.98% iron sulfate, 50 g NaCl, 1 L distilled water at pH 7.4 [20]). After 7 days cultivation at 28°C on a rotary shaker at 200 rpm, 0.5 mL of the seed culture was used to inoculate 50 mL of CM, CA02LB (20 g mannitol, 20 g soybean meal, 1 L distilled water at pH 7.5) or CA07LB (15 g Glycerol, 30 g oatmeal, 5 g yeast extract, 5 g KH2PO4, 5 g Na2HPO4.12H2O, 1 g MgCl2.6H2O, 1 L distilled water at its natural pH) in 250 mL Erlenmeyer flasks. All media was supplemented with 5-10% NaCl (w/v) when necessary. The fermentation was carried out for 7 days at 28 °C at 200 rpm. Chemicals and reagents for media preparation were purchased from Sigma.
Extraction and isolation
For the initial chemical analysis the entire 50 ml cultures of N. kunsanensis grown in various media were freeze-dried before extracting with methanol for LCMS analysis. For the scale up fermentation for compound isolation, the cultures (80 × 50 mL, total 4 L) of N. kunsanensis from all flasks were first combined and centrifuged to separate the culture supernatant and the mycelia. The combined mycelia were freeze-dried, shaken overnight 2 times with MeOH/ CH2Cl2 (4 L), filtered and added to 1 L of water. The organic layer from the solvent partition was evaporated to dryness using rotary evaporation. The dried dichloromethane extract was dissolved in DMSO and separated by C18 reversed-phase preparative HPLC (solvent A: H2O + 0.1% HCOOH, solvent B: ACN + 0.1% HCOOH; flow rate: 18 mL/min, gradient conditions:70:30 isocratic for 5 minutes; 30% to 40% of solvent B over 5 minutes, followed by 40% to 70% of solvent B over 40 minutes, and an increase from 70% to 100% of solvent B over 50 minutes, and to 100% solvent B in 15 minutes) to give 0.2 mg of anthracimycin (1) with impurities and 0.9 mg of anthracimycin BII-2619 (2).
Chemical Structural Data
The NMR spectra of the compounds are provided in Supplementary Figures S1 (S1a-S1d) to S2 (S2a-S2f). Below, we list the chemical structural data including the measured molar circular dichroism (Δε).
Anthracimycin standard
Amorphous white powder; [α]D -7 (c 2.93, CH2Cl2); UV (MeOH) λmax (log ε) 235 (4.21) and 286 (3.76) nm; ECD (MeOH) λ (Δε) 202 (+40.47), 214 (+12.96), 236 (+34.84), 283 (-28.84); HRESIMS m/z 397.2374 [M+H]+ (calculated for C25H32O4, 397.2379).
Anthracimycin BII-2619 (2)
Amorphous white powder; [α]D -47 (c 0.16, CH2Cl2); UV (MeOH) λmax (log ε) 235 (4.17) and 286 (3.62) nm; ECD (MeOH) λ (Δε) 201 (+30.64), 213 (+7.53), 236 (+18.14), 283 (-15.98); HRESIMS m/z 397.2371 [M+H]+ (calculated for C25H32O4, 397.2379).
Analytical Chemistry Procedures
Optical rotations were recorded on a JASCO P-2000 digital polarimeter. Electronic circular dichroism (ECD) was measured on a JASCO J815 - circular dichroism (CD) spectrometer. The CD spectra of all samples were recorded in methanol at molar concentration of 0.025 mM in 195-400 nm region. UV spectra were obtained on a General Electric Ultrospec 9000 spectrophotometer.
NMR spectra were collected on a Bruker DRX-400 NMR spectrometer with Cryoprobe, using 5-mm BBI (1H, G-COSY, multiplicity-edited G-HSQC, and G-HMBC spectra) or BBO (13C spectra) probe heads equipped with z-gradients. Spectra were calibrated to residual protonated solvent signals (CHCl3 δH 7.24 and CDCl3 δC 77.23).
The HRESIMS and MS/MS spectra were acquired on Agilent UHPLC 1290 Infinity coupled to Agilent 6540 accurate-mass quadrupole time-of-flight (QTOF) mass spectrometer equipped with a splitter and an ESI source. The analysis was performed with a C18 4.6 x 75 mm, 2.7 µm column at flowrate of 2 mL/min, under standard gradient condition of 100% water with 0.1% formic acid to 100% acetonitrile with 0.1% formic acid over 15 minutes. The chiral analysis was performed on a chiral column Lux® 3 µm amylose-1 at flowrate of 0.4 mL/min, under gradient condition of 75% to 100% acetonitrile/water with 0.1% formic acid over 6.5 minutes.
The typical QTOF operating parameters were as follows: positive ionization mode; sheath gas nitrogen, 12 L/min at 295 °C; drying gas nitrogen flow, 8L/min at 275 °C; nebulizer pressure, 30 psig; nozzle voltage, 1.5 kV; capillary voltage, 4 kV. Lock masses in positive ion mode: purine ion at m/z 121.0509 and HP-921 ion at m/z 922.0098. MS/MS data of precursor ions were acquired using collision energies between 10-40 eV and acquisition rate at 2 spectra/s.
Preparative HPLC was performed on the Agilent 1260 Infinity Preparative-Scale LCMS Purification System, completed with Agilent OpenLAB CDS ChemStation Edition for LC and LCMS Systems. Anthracimycin and analogues were separated on an Agilent Prep C18 column (100 × 30 mm) by gradient elution with a mixture of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B).
Bacterial whole cell assays
MIC was determined according to guidelines of the Clinical Laboratory Standards Institute (CLSI) [19] with slight modifications using the broth dilution method in a 384-well microplate format. Inoculum of each test organism was prepared by resuspending isolated colonies in sterile saline to a 0.5 McFarland standard. An inoculum of 5 x 105 CFU/mL was incubated with the compounds for 24 hours at 37°C. The effect of the compounds on bacterial growth was evaluated by measuring the optical density at 600 nm using a microplate reader.
Mammalian cell cytotoxicity
A549 human lung carcinoma cells seeded at 1500 cells/well in a 384-well microplate were treated with the compounds for 72 hours at 37°C in the presence of 5% CO2. PrestoBlue™ cell viability reagent (Life Technologies) was used to assess the cytotoxic effect of the compounds. Microplates were incubated with this dye for 2 hours before fluorescence reading at excitation 560 nm and emission 590 nm.
Results
Predicting the atc biosynthetic gene cluster in a different organism
The published atc biosynthetic gene cluster in Streptomyces sp. T676 comprises nine genes/proteins (AtcA-AtcI, accession no. LN871452), where four of them (AtcC-AtcF) have been proposed to be functionally involved in the generation of anthracimycin. These four proteins can be split into 10 Polyketide Synthetase (PKS) modules, and each module harbours specific functional domains required for their molecular function [1].
As a result of a blast/psi-blast search [15] with all functional domains described in the Streptomyces sp. T676 atc cluster against nr [16], we generated a list of organisms harbouring all or almost all of these domains (E-value cut-off 0.001, see Methods). As expected, we retrieved the four Streptomyces strains discussed above first but, in addition, we found sequence equivalents for all 41 functional domains for AtcC-AtcF in the genome of Nocardiopsis kunsanensis, a species belonging to the same taxonomic class (Actinobacteria) but in a different order (Streptosporangiales) compared to Streptomyces (Supplementary Table S1). The next best hit is the species Sorangium cellulosum missing one domain from module 9. Interestingly, Sorangium cellulosum is known to produce chlorotonil A, a compound with a scaffold very similar to that of anthracimycin.
As a next step, we tried to understand whether the detected functional domains in N. kunsanensis are sequentially organized in a cluster and, if so, whether this cluster resembles the atc biosynthetic cluster. Tblastn searches with the annotated T676 functional protein domains as queries against N. kunsanensis contigs were carried out. All functional domains had a significant hit and could be mapped to N. kunsanensis DSM 44524 contig 32 (accession no. NZ_ANAY01000032) (Figure 2). Furthermore, blast searches with the four Streptomyces sp. T676 atc (AtcC-AtcF) proteins (accession no. CTQ34880, CTQ34881, CTQ34882, CTQ34883, respectively) against the N. kunsanensis proteome produced significant hits (E-value = 0.0 for all of them) to four N. kunsanensis proteins: WP_017577639 (60% sequence identity to AtcC), WP_017577638 (50% sequence identity to AtcD), WP_017577637 (54% sequence identity to AtcE), and WP_020480252 (55% sequence identity to AtcF). For all results, the sequence alignment coverage was ≥ 98% (Supplementary Table S2). Interestingly, WP_017577639 is annotated as [acyl-carrier-protein] S-malonyltransferase; the other three proteins are annotated as hypothetical proteins.
Finally, we tested the detected N. kunsanensis contig 32 (NZ_ANAY01000032) with antiSMASH [21]. The result clearly supports the existence of an anthracimycin producing trans-AT PKS cluster in N. kunsanensis (Supplementary Figure S3). Notably, the N. kunsanensis NZ_ ANAY01000032 cluster was not part of the antiSMASH gene cluster database when this study was carried out in December 2015.
Based on the alignment of the four AtcC, AtcD, AtcE and AtcF proteins of the four Streptomyces strains discussed above and N. kunsanensis, a phylogenetic tree was created (Figure 3). It shows that the four Streptomyces strains cluster together, and the N. kunsanensis cluster sequence is the outlier, being the most distant one. This is also true for each single PKS module, with the Nocardiopsis sequences showing the lowest sequence identity to Streptomyces sp. T676 compared to all other Streptomyces studied (Supplementary Figure S4). But the inspection of core regions of the critical functional domains, keto-synthase (KS), dehydratase (DH), ketoreductase (KR), methyltransferase (MT), acyl carrier protein (ACP) and thioesterase (TE) (see Figure 5 in [1]), shows that all critical amino acid residues are strongly conserved among all four Streptomyces strains and N. kunsanensis (Supplementary Figure S5a).
To conclude, in December 2015 we suggested that the genome of N. kunsanensis carries an atc trans-AT PKS biosynthetic cluster. It appears to contain all functional domains compared to the known Streptomyces atc cluster, and the critical residues in the core regions of all ACP domains, DH domains, KR domains, KS domains, MT domains, and TE domains are present. The sequence of the N. kunsanensis atc cluster is the most distant one compared to the four available anthracimycin producing Streptomyces strains, nevertheless, we speculated that N. kunsanensis is probably able to produce anthracimycin or its derivatives despite the evolutionary divergence from the Streptomyces genus.
Experimental validation of anthracimycin derivative production by N. kunsanensis
In order to experimentally validate the production of anthracimycin or its derivative by N. kunsanensis, following the identification of the presence of the respective biosynthetic gene cluster in its genome, we analysed the methanol extracts derived from the a small scale 50 mL fermentation of N. kunsanensis in 3 different media, CM, CA02LB and CA07LB. For this initial analysis, we freeze-dried the entire 50 mL cultures and extracted the metabolites with methanol. The methanol extracts were then analysed with ultra-performance liquid chromatography (UPLC), combined with quadrupole-time of flight high resolution mass spectrometry under positive ionization (ESI) mode.
The extracts were screened for the precursor ion of anthracimycin (m/z 397.2379) and matched against an authentic anthracimycin standard (1) isolated by our group from the Streptomyces sp. T676 [1] (retention time of 13.4 min, Supplementary Figure S6a). The extracted ion chromatogram (EIC) of m/z 397.2379 showed that N. kunsanensis produced the highest amount of the metabolite in CM (Supplementary Figure S6e); however, the retention time for the peak was observed at 13.2 min. The observed difference of 0.2 min is one order of magnitude higher than the retention drift in our LC/QTOF system suggesting the production of a compound that is different from anthracimycin, but most likely its derivative. Furthermore, close examination of its MS/MS spectra showed identical fragment ions to those of anthracimycin standard (1) at higher mass range (m/z > 311) but with a difference of ~14.02 Da at lower mass range (Supplementary Figures S7a-c and S8a-c). The fragmentation patterns suggested that the major compound from N. kunsanensis is a new analogue of anthracimycin with structural difference in a methyl position.
A 4 liter fermentation of N. kunsanensis in complex medium (CM) was conducted with the aim of isolating sufficient amount of the new analogue to determine its structure. After seven days of cultivation a small aliquot of the cell biomass was separated from the broth and both were extracted with methanol for analysis by LCMS. The extracted ion chromatogram of the biomass extract showed a major peak at 13.2 min for the potential new analogue of anthracimycin as well as a minor peak at 13.4 min for anthracmycin (1) (Supplementary Figure S6a). To note, the expected error rate for HPLC device is clearly below 0.05 min [22]. Only trace amount of anthracimycins were detected in the broth (data not shown).
The biomass collected from the 4 liter fermentation was then freeze-dried before extracting with dichloromethane/methanol (1:1) and partitioned with water. The dichloromethane fraction was dried, dissolved in DMSO and fractionated by reverse phase preparative HPLC to afford a suite of compounds. A new anthracimycin analogue, anthracimycin BII-2619 (2) was isolated together with a minor amount of the known compound anthracimycin (1). The structure of anthracimycin was determined by 1H NMR and LCMS/MS experiments (Supplementary Figure S7), whereas that of anthracimycin BII-2619 was elucidated by 1D and 2D NMR as well as LCMS/MS experiments (Supplementary Figure S8).
In order to further ascertain that 0.2 min difference in retention time is not a measurement error between anthracimycin (1) and anthracimycin BII-2619 (2), the three NMR samples (anthracimycin (1), anthracimycin standard and anthracimycin BII-2619 (2)) were re-analyzed using an isocratic gradient. Extracted ion chromatogram of m/z 397.2379 (Supplementary Figure S6b) showed anthracimycin (1) and anthracimycin standard at retention time 3.6 min, whereas anthracimycin BII-2619 (2) is indeed clearly different at 3.2 min.
Anthracimycin (1) was obtained from the 4 liter fermentation of N. kunsanensis with impurities, but was found to have a comparable 1H NMR spectra (Table 1, Supplementary Figure S1a and b) as our anthracimycin standard that we purified from Streptomyces sp. T676. The retention time, UV absorption spectrum and MS/MS fragmentation patterns of these two sources of anthracimycin on a chiral column (Lux® 3 µm amylose-1) were identical (Supplementary Figure S9). The electronic circular dichroism (ECD) results for our anthracimycin standard (Supplementary Figure S10) were in good agreement with those of the recently reported anthracimycin (Supplementary page S20 in ref. [4]). This suggests that the anthracimycin produced by N. kunsanensis and Streptomyces sp. T676 are of the same chirality as that produced by Streptomyces sp. CNH365. The full structure and absolute configuration at all asymmetric carbon centers of anthracimycin (1) have been assigned by Jang et al. [4] on the basis of X-ray and NMR analysis. Hence, the structure of anthracimycin (1) was defined as depicted in Figure 1.
NMR spectral dataa of anthracimycin (1) and anthracimycin BII-2619 (2)
| Position | 1 | 2 | |||
|---|---|---|---|---|---|
| 13Cb | 1Hc, mult. (J = Hz) | 13Cb | 1Hc, mult. (J = Hz) | ||
| 1 | 168.9 | - | 165.7 | - | |
| 2 | 49.3 | 3.50, q (7.0) | 46.5 | 3.47, d (11.5) | |
| 3.21, d (11.5) | |||||
| 3 | 190.9 | - | 184.3 | - | |
| 4 | 103.0 | 5.96, s | 102.6 | 5.96, s | |
| 5 | 194.3 | - | 197.9 | - | |
| 6 | 52.8 | 2.58, m | 49.7 | 2.72, dd (11.8, 6.7) | |
| 7 | 33.1 | 1.95, m | 35.5 | 2.08, m | |
| 8α | 31.4 | 2.39, br d (16.0) | 30.9 | 2.59, m | |
| 8β | 1.52, m | ||||
| 9 | 121.0 | 5.36, m | 128.3 | 5.34, m | |
| 10 | 134.0 | - | 132.5 | - | |
| 11α | 37.6 | 2.02, m | 38.3 | 1.99, dd (16.5, 4) | |
| 11β | 1.82, dd (16.5, 10.3) | 1.72, m | |||
| 12 | 37.5 | 1.95, m | 35.5 | 2.10, m | |
| 13 | 133.1 | 5.71, d (10) | 133.7 | 5.73, d (10) | |
| 14 | 125.0 | 5.53, m | 124.4 | 5.52, m | |
| 15 | 46.1 | 2.60, m | 45.8 | 2.64, m | |
| 16 | 33.1 | 2.64, m | 33.1 | 2.62, m | |
| 17 | 139.1 | 5.40, m | 139.2 | 5.37, m | |
| 18 | 126.2 | 5.87, br t (11) | 126.1 | 5.86, br t (11) | |
| 19 | 123.9 | 6.45, br ddd (15, 11, 2) | 123.9 | 6.46, br ddd (15, 11, 2) | |
| 20 | 131.7 | 5.56, m | 131.6 | 5.54, m | |
| 21 | 70.0 | 5.57, m | 70.2 | 5.55, m | |
| 22 | 21.0 | 1.33, d (6.5) | 21.0 | 1.33, d (6.5) | |
| 23 | 11.9 | 1.39, d (7) | - | - | |
| 24 | - | - | 15.4 | 0.73, d (7.0) | |
| 25 | 23.6 | 1.67, s | 23.6 | 1.65, s | |
| 26 | 16.5 | 0.94, d (7) | 16.5 | 0.92, d (7) | |
a Assignments based on COSY, HSQCED and HMBC. b (1) and (2) were recorded at 100 MHz with CDCl3 as internal standard at δ 77.23, Chemical shifts (δ) in ppm. c (1) and (2) was recorded at 400 MHz with CDCl3 as internal standard at δ 7.24,. s: singlet; d: doublet. q: quartet; m: multiplet; br; broad; Chemical shifts (δ) in ppm. The 1H and 13C NMR data of (1) and (2) are very similar except for the presence of an isolated methylene group. Relevant positions are highlighted in bold.
Anthracimycin BII-2619 (2) was purified as a white powder and was found to have the same molecular formula C25H32O4 and UV as anthracimycin (1). As we have much less than 10mg of the compound, X-ray crystallographic structural determination, the most reliable approach, is not possible. Our structural proposal (2) resides on the following arguments:
- The MS/MS spectra showed characteristic fragment ions of anthracimycin (1) at m/z 379.2272 [M-H2O+H]+, 351.2317 [M-HCOOH+H]+, 329.1751 [M-C5H7+H]+ and 311.1645 [M-C5H9O+H]+. However, the lower mass range at m/z 269.1540, 205.0861 and 107.0858 showed a difference of 14.0159 Da to that of anthracimycin (1) (m/z 255.1383, 219.1020 and 93.0699 in Supplementary Figures S7a-c and S8a-c). Thus, the new compound has the same upper MS/MS spectrum as anthracimycin but a difference by one methyl-group mass in lower end. Thus, the most likely reason for a structural difference is the changed position of a methyl group. The fragmentation patterns suggested a change in the methyl position, possibly from C-2 to C-8 in (2) as compared to (1).
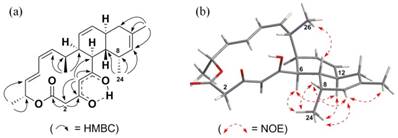
- The 1D 1H and 13C NMR data and the 2D COSY, HSQC and HMBC data for the new compound are very similar to that of anthracimycin except for that of a differently placed methyl group. Therefore, we conclude that the structural frame of the new compound repeats the structure of anthracimycin which is known from X-ray crystallography. The 1H and 13C NMR data of (1) and (2) were very similar except for the presence of an isolated methylene group at δH 3.21 (d, J 11.5 Hz), 3.47 (d, J 11.5 Hz) and a methyl signal at δH 0.73 (3H, d, J 7 Hz) in the 1H NMR spectrum of (2). The isolated methylene was positioned at C-2, in between an enolised β-diketone and a lactone moiety, and was supported by the 2JCH and 3JCH HMBC correlations (Figure 4) from H-2 to C-1, H-2 to C-3 and H-4 to C-2.
- The HMBC data indicates that the methyl group shifted from C-2 to C-8: The observed methyl signal at δH 0.73 was assigned to C-24 attached to C-8, based on the evidence provided by the 2JCH and 3JCH HMBC correlations from H-24 to C-7, H-24 to C-8, and H-24 to C-9. The deduced structure and the relative configurations of (2) are entirely consistent with the rest of the HMBC and the NOESY correlations (Figure 4, Supplementary Figures S2e and S2f).
- The absolute configuration of anthracimycin BII-2619 (2) was assumed to be the same as anthracimycin (1), except C-24-methyl, based on the similarities in their 1H and 13C chemical shifts as well as (a) due the essentially identical ECD spectra compared with that of the anthracimycin standard (they really pinpoint the chirality adjacent to the chromophore β-diketone, see Supplementary Figure S10) [23] and (b) the absence of an enantiodivergence step in their biosynthesis. Thus, the absolute configuration of anthracimycin BII-2619 (2) at positions 6, 7, 12, 15, 16, 21 were assumed to be the same as anthracimycin (1) [4], based on the similarities in their 1H and 13C chemical shifts, coupling constants and no enantiondivergence step in their biosynthesis.
- Therefore, we can interpret the NOESY data for deriving the relative spatial position of C-24 methyl attached to C-8. The α orientation of the C-24 methyl group was deduced by the unusual shielding effect observed for the C-24 methyl group at δH 0.73 (3H, d, J 7 Hz), which is due to the anisotropy effect from the proximate C-5 ketone function, and is only possible if the C-24 methyl group is in the α position. This was further supported by the observed H-24/H-6 and H-24/H-12 NOESY correlations (Supplementary Figure S2f). In fact, the spectral data which contains the C-24 methyl group of anthracimycin BII-2619 (2) resemble those of chlorotonil A (3) [8,9]. Hence, the structure of anthracimycin BII-2619 (2) was established as shown in Figure 1.
Key HMBC and NOE correlations of anthracimycin BII-2619 (2). The arrows indicate (a) the coupling observed between the hydrogens and carbons in the HMBC and (b) the NOE observed between the hydrogens in the NOESY.
To emphasize, although anthracimycin BII-2619 (2) was the major product purified, N. kunansensis does produce also minor amounts of classic anthracimycin (1) as well as even lesser amounts of other analogues (data not shown). Both the 1H NMR (Supplementary Figure S1a) and EIC (Supplementary Figure 6a) showed that classic anthracimycin (1) isolated from N. kunsanensis was not pure, due to its low abundance. To make the 1H NMR data derived from the anthracimycin standard as well as from anthracimycin (1) and anthracimycin BII-2619 (2) purified from N. kunsanensis more comparable, we show the data fused into one figure with x-axis (Figure S1d). Indeed, the 1H data of (1) and (2) are very similar except for the presence of an isolated methylene group at δ 3.21 (d, J 11.5 Hz), 3.47 (d, J 11.5 Hz) (see left box in Figure S1d) and a methyl signal at δ 0.73 (3H, d, J 7 Hz) in the 1H NMR spectrum of (2) (see right box in Figure S1d, lane (c)). Thus, the quartet signal is seen only in lanes (a) and (b) of Figure S1d (anthracimycin standard and anthracimycin from N. kunansensis) but not in lane (c) (anthracimycin BII-2619).
In summary, Figure S1d shows the 8-methyl signal (position C-24) at δ 0.73 ppm (right box, lane (c)) for anthracimycin BII-2619 and the 2-methyl signal (position C-23) at δ 1.39 ppm for anthracimycin (1) and anthracimycin standard (lanes (a) and (b), respectively). These results suggest that the methyl signal is absent in anthramycin (1) and anthracimycin standard 1H NMR spectrum. Thus, the LCMS and NMR data have unambiguously shown that N. kunsanensis produces both anthracimycin and anthracimycin BII-2619.
Biological activities
Antibacterial activity of anthracimycin BII-2619 (2) was assessed against methicillin-sensitive and -resistant Staphylococcus aureus (MSSA and MRSA) and compared with that of anthracimycin standard. Both compounds showed minimum inhibitory concentration (MIC) values of ≤1μM, and were thus further profiled against other bacterial cultures and a human laboratory cancer cell line (Table 2). Anthracimycin and anthracimycin BII-2619 (2) displayed activity against Bacillus subtilis and Enterococcus faecalis but were inactive against the tested Gram-negative bacteria. Anthracimycin was, however, still more potent against Enterococcus faecalis (~60-fold) and Bacillus subtilis (~100-fold) when compared to anthracimycin BII-2619 (2). Both compounds demonstrated moderate, very similar low cytotoxic activity.
Biological Activity of anthracimycin standard and anthracimycin BII-2619 (2)
| Target Organism or Cell line | Activitya (μg/ml [μM]) | |
|---|---|---|
| (1) | (2) | |
| Gram Positive Bacteria | ||
| MRSA (ATCC 33591) | 0.06 [0.15] | 0.125 [0.32] |
| MSSA (ATCC 25923) | 0.06 [0.15] | 1 [2.5] |
| Bacillus subtilis (ATCC 6633) | 0.5 [1.3] | 64 [161] |
| Enterococcus faecalis (ATCC 29212) | 0.125 [0.32] | 8 [20] |
| Gram Negative Bacteria | ||
| Escherichia coli (ATCC 25922) | >128 [>323] | >128 [>323] |
| Pseudomonas aeruginosa (ATCC 27853) | >128 [>323] | >128 [>323] |
| Mammalian Cells | ||
| A549 Human lung carcinoma cells (ATCC CCL-185) | ~ 32 [~81] | 35 [88] |
a IC90 for bacterial assays and IC50 for mammalian cell assay. All assays were performed in triplicate on two different test runs.
Discussion
The need for the identification of novel antibiotics in order to deal with the increasing problem of drug-resistance development has led to a continuous increase in the search for natural products [24]. As most antibiotics are derived from natural products [25], this research area will greatly benefit from -omics technologies since mining of genome sequences in silico will efficiently generate lists of organisms with the potential ability to synthesize compounds of desired structure classes [24,26]. The respective shortcuts will then direct experimental validation efforts and, thus, save valuable time and cost. This approach was applied in the present work, as we screened the existing genomic data to find organisms that carry sequences similar to the atc biosynthetic cluster of Streptomyces. For example, we found in our analysis that Streptomyces sp. NRRL F-5065 carries the atc biosynthetic gene cluster; yet, it is the only reported strain not experimentally tested to produce anthracimycin. We were surprised to also hit Nocardiopsis kunsanensis, a distant relative of Streptomyces from another taxonomic order.
Occasionally observing similar or identical genes or even gene clusters in diverse genera as a consequence of horizontal gene transfer is to be expected in the context of large scale comparative genomics efforts [27-29]. Nevertheless, it is interesting to see the atc biosynthetic gene cluster outside the Streptomycetales phylogenetic branch as an additional example of a trans-AT PKS cluster that could potentially evolve by this mode of action.
The identification of alternative organisms producing any known compound is of importance on different levels. For instance, they can lead to the discovery of new major compounds that are analogues of the existing ones. This has been the case here with the identification of compound anthracimycin BII-2619 produced by N. kunsanensis. This new analogue automatically becomes a candidate compound for further testing.
Our experimental results prove that N. kunsanensis produces the derivative anthracimycin BII-2619 as the major compound and anthracimycin in minor quantities. This finding further exemplifies the exceptional importance of Actinobacteria for natural product discovery [26], serving as potential source of chemical agents against different drug-resistant infections [25], for refactoring synthetic pathways [30], as well as for finding unique chemical structures that may form the scaffold of novel drugs [31]. In addition, knowing more than one organism that produce the same compound can be of crucial help to evolutionary studies on the origin of their biosynthetic gene clusters.
The only chemical difference observed between anthracimycin (1) and anthracimycin BII-2619 (2) is the presence of methyl groups at the C-2 and C-8 positions, respectively (Figure 1). In total, anthracimycin and its derivative carry up to four methyl groups at C-2, C-8, C-10 and C-16. Since the atc cluster contains only three methyltransferases (MT3, MT7, and MT10; Figure 2) and these methyltransferases are implied to catalyze the methylation at C-16, C-10 and C-2 respectively [1], we have to consider that methylation at C-8 is possibly carried out by either a post-PKS tailoring reaction catalyzed by a methyltransferase that is external to the atc PKS cluster, by MT7 if it is to assume its significant deviation from colinearity and possible cross-modular function as described by Alt and Wilkinson [1], or by MT10, due to its C-terminal localization that could help it to function as a post-PKS tailoring enzyme. There are currently >20 proteins from N. kunsanensis labelled as S-adenosylmethionine- (SAM-) dependent methyltransferases out of >70 annotated methyltransferases. Attempts to identify the actual enzyme for this reaction would require an extensive effort that is out of the scope of this work. Yet, at this point, alternative scenarios cannot be excluded such as some PKS cluster methyltransferases being less than 100% efficient or with methylation site promiscuity.
To note, a similar hypothesis was already proposed for the chlorotonil A biosynthesis. The PKS domain organization of chlorotonil A producing strain S. cellulosum 1525 is different from the anthracimycin producing Streptomyces and N. kunsanensis. In this case, the methyltransferases MT3, MT6, and MT7 of the PKS cluster were implied to be responsible for the methylation at C-16, C-8, and C-10, respectively, whereas facultative methylation at C-2 was accredited to the free-standing SAM-dependent methyltransferase CtoF (accession no. ALD83694.1), which is not part of the PKS cluster [9]. The final answer to this question will require a dedicated analytical experiment.
We wish to emphasize that the structure of anthracinycin BII-2619 (2) as well as those of anthracimycin (1) and chlorotonil A also cannot be strictly explained based on the type I PKS organization, that several significant deviation from collinearity of enzymes have been described and, generally, that the co-linearity rule is only well applied to type I PKS with cis-AT organization [1,2,9]. The biosynthetic gene cluster reported here falls into the trans-AT PKS classification and so do the anthracimycin (1) and chlorotonil A clusters as well. Importantly, the biosynthetic gene cluster for chlorotonil A was shown to also produce new chlorotonil congeners that additionally exhibit carbon skeleton variations, which illustrates the partial promiscuity of these megaenzymes and the difficulty to ascertain all reaction steps to individual modules and the assignments of functions in the previous well cited work as well as in our new one remains tentative.
One of their scenarios to explain such differences is the inefficient methylation functionality in module 7. Although it is true that we used the genome of N. kunsanensis DSM 44524, which by definition is a draft genome, one should be aware that current sequencing and assembly technology produces draft genomes that cover 95-99% of the full genome in the case of bacteria, as benchmarked in GAGE [32] and in cases of lower coverage, such as for Rhodobacter sphaeroides, the missing regions came from short low complexity repeat regions that are difficult to assemble but do not harbor biosynthetic gene clusters.
Still, we ran antiSMASH against the complete draft genome of N. kunsanensis to find all its predicted biosynthetic gene clusters. We were able to identify the cluster here reported as the only trans-AT PKS. To compare this cluster to other predicted PKS types of clusters, we extracted all the predicted KS domains, aligned and calculated their phylogenetic distances as in the Figure S5b. This tree is in perfect agreement with the Figure S16 in suppl. material of reference [9] allowing to identify clades of KS domains based on their substrate specificity. This tree shows that all other PKS clusters found in the genome are not closely related to the one producing anthracimycin BII-2619, which itself remains closely related to the one producing chlorotonil A.
We also compared the biological activity of anthracimycin standard and anthracimycin BII-2619 (2). We found that both compounds were effective against Gram-positive bacteria, but anthracimycin BII-2619 is 50- to 100-fold less active compared to anthracimycin. Both compounds are inefficient for suppressing the growth of Gram-negative microbes and have similar level of toxicity against the lung cancer cell line A549. Low cytotoxicity was also observed for HeLa cells [10]; however it was recently reported that anthracimycin was approximately 10-fold more toxic against three human hepatocellular carcinoma cell lines [33]. Thus, the toxicity of anthracimycin and its analogs needs to be further assessed against more cell types. As stated above, the only structural difference between these two compounds is the presence of methyl groups at the C-2 in anthracimycin (1) and C-8 position for anthracimycin BII-2619 (2). This shift of the methyl group from the C-2 to C-8 position appeared to have no effect on the cytotoxicity of this compound on A549 cells, but significantly decreased its antibacterial potency.
To conclude, we report the anthracimycin derivative producing biosynthetic gene cluster in N. kunsanensis that was identified through genome sequence screening. We show that this computational approach is able to identify different organisms that produce the same or similar compounds. It is the first time that the atc biosynthetic cluster has been reported for another order rather than Streptomycetales. We were able to prove that this strain also produces anthracimycin in small quantities under specific fermentation conditions. The identification of its main product anthracimycin BII-2619 (2) together with its bioactivity tests contribute to the understanding of anthracimycin as a potential candidate antibiotic.
Supplementary Material
Supplementary figures and tables.
Abbreviations
atc: anthracimycin; NMR: nuclear magnetic resonance; HMBC: heteronuclear multiple bond correlation; COSY: homonuclear correlation; HSQCED: heteronuclear single quantum coherence - editing: NOESY: Nuclear Overhauser Effect Spectroscopy.
Acknowledgements
This work was supported by the Agency for Science, Technology and Research (A*STAR), Singapore.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Alt S, Wilkinson B. Biosynthesis of the Novel Macrolide Antibiotic Anthracimycin. ACS Chem Biol. 2015;10:2468-79
2. Helfrich EJN, Piel J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat Prod Rep. 2016;33:231-316
3. Nguyen T, Ishida K, Jenke-Kodama H, Dittmann E, Gurgui C, Hochmuth T. et al. Exploiting the mosaic structure of trans-acyltransferase polyketide synthases for natural product discovery and pathway dissection. Nat Biotechnol. 2008;26:225-33
4. Jang KH, Nam S-J, Locke JB, Kauffman CA, Beatty DS, Paul LA. et al. Anthracimycin, a potent anthrax antibiotic from a marine-derived actinomycete. Angew Chem Int Ed Engl. 2013;52:7822-4
5. Doroghazi JR, Albright JC, Goering AW, Ju K-S, Haines RR, Tchalukov KA. et al. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol. 2014;10:963-8
6. Harunari E, Komaki H, Igarashi Y. Biosynthetic origin of anthracimycin: a tricyclic macrolide from Streptomyces sp. J Antibiot (Tokyo). 2016;69:403-5
7. Komaki H, Ichikawa N, Hosoyama A, Fujita N, Harunari E, Igarashi Y. Draft Genome Sequence of an Anthracimycin Producer, Streptomyces sp. TP-A0875. Genome Announc. 2015:3
8. Gerth K, Steinmetz H, Höfle G, Jansen R. Chlorotonil A, a macrolide with a unique gem-dichloro-1,3-dione functionality from Sorangium cellulosum, So ce1525. Angew Chem Int Ed Engl. 2008;47:600-2
9. Jungmann K, Jansen R, Gerth K, Huch V, Krug D, Fenical W. et al. Two of a Kind-The Biosynthetic Pathways of Chlorotonil and Anthracimycin. ACS Chem Biol. 2015;10:2480-90
10. Hensler ME, Jang KH, Thienphrapa W, Vuong L, Tran DN, Soubih E. et al. Anthracimycin activity against contemporary methicillin-resistant Staphylococcus aureus. J Antibiot (Tokyo). 2014;67:549-53
11. Held J, Gebru T, Kalesse M, Jansen R, Gerth K, Müller R. et al. Antimalarial activity of the myxobacterial macrolide chlorotonil a. Antimicrob Agents Chemother. 2014;58:6378-84
12. Weissman KJ. The structural biology of biosynthetic megaenzymes. Nat Chem Biol. 2015;11:660-70
13. Eisenhaber B, Kuchibhatla D, Sherman W, Sirota FL, Berezovsky IN, Wong W-C. et al. The Recipe for Protein Sequence-Based Function Prediction and Its Implementation in the ANNOTATOR Software Environment. Methods Mol Biol. 2016;1415:477-506
14. Sirota FL, Ooi H-S, Gattermayer T, Schneider G, Eisenhaber F, Maurer-Stroh S. Parameterization of disorder predictors for large-scale applications requiring high specificity by using an extended benchmark dataset. BMC Genomics. 2010;11(Suppl 1):S15
15. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389-402
16. Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2015;43:D30-35
17. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772-80
18. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725-9
19. CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard-Ninth Edition. CLSI Document M07- A9; Wayne, PA: Clinical and Laboratory Standards Institute. 2012
20. Chun J, Bae KS, Moon EY, Jung SO, Lee HK, Kim SJ. Nocardiopsis kunsanensis sp. nov, a moderately halophilic actinomycete isolated from a saltern. Int J Syst Evol Microbiol. 2000;50(Pt 5):1909-13
21. Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R. et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237-243
22. Dolan JW. How Much Retention Time Variation Is Normal? [cited 2018 Apr 8]. Available from: http://www.chromatographyonline.com/how-much-retention-time-variation-normal-0
23. Li X-C, Ferreira D, Ding Y. Determination of Absolute Configuration of Natural Products: Theoretical Calculation of Electronic Circular Dichroism as a Tool. Curr Org Chem. 2010;14:1678-97
24. Zhou Z, Gu J, Du Y-L, Li Y-Q, Wang Y. The -omics Era- Toward a Systems-Level Understanding of Streptomyces. Curr Genomics. 2011;12:404-16
25. Abdelmohsen UR, Balasubramanian S, Oelschlaeger TA, Grkovic T, Pham NB, Quinn RJ. et al. Potential of marine natural products against drug-resistant fungal, viral, and parasitic infections. Lancet Infect Dis. 2017;17:e30-41
26. Gomez-Escribano JP, Alt S, Bibb MJ. Next Generation Sequencing of Actinobacteria for the Discovery of Novel Natural Products. Mar Drugs. 2016:14
27. Doroghazi JR, Buckley DH. Widespread homologous recombination within and between Streptomyces species. ISME J. 2010;4:1136-43
28. Doroghazi JR, Metcalf WW. Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genomics. 2013;14:611
29. Jensen PR. Natural Products and the Gene Cluster Revolution. Trends Microbiol. 2016;24:968-77
30. Tan G-Y, Liu T. Rational synthetic pathway refactoring of natural products biosynthesis in actinobacteria. Metab Eng. 2017;39:228-36
31. Manivasagan P, Kang K-H, Sivakumar K, Li-Chan ECY, Oh H-M, Kim S-K. Marine actinobacteria: an important source of bioactive natural products. Environ Toxicol Pharmacol. 2014;38:172-88
32. Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S. et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012;22:557-67
33. Hayashi T, Yamashita T, Okada H, Oishi N, Sunagozaka H, Nio K. et al. A Novel mTOR Inhibitor; Anthracimycin for the Treatment of Human Hepatocellular Carcinoma. Anticancer Res. 2017;37:3397-403
Author contact
![]() Corresponding authors: Email: fernandaa-star.edu.sg, frankea-star.edu.sg, yoganathanka-star.edu.sg, ngsba-star.edu.sg
Corresponding authors: Email: fernandaa-star.edu.sg, frankea-star.edu.sg, yoganathanka-star.edu.sg, ngsba-star.edu.sg