ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2018; 6:34-40. doi:10.7150/jgen.22460 This volume Cite
Research Paper
In-silico Designing and Testing of Primers for Sanger Genome Sequencing of Dengue Virus Types of Asian Origin
Ajay Prakash Joshi1, Annette Angel1, Bennet Angel1,2, Rajendra Kumar Baharia1,3, Suman Rathore1,4, Neha Sharma1, Karuna Yadav1, Sharad Thanvi5, Indu Thanvi6, Vinod Joshi1,2 ![]()
1. Desert Medicine Research Centre, Indian Council of Medical research, Jodhpur, India-342005
2. Present Address: Amity Institute of Virology & Immunology (AIVI), Amity University, Noida, U.P., India- 201313
3. Present Address: National Institute of Malaria Research, Secotr-8, Dwarika, New Delhi -110077
4. All India Institute of Medical Sciences, Jodhpur, India
5. Department of Neuroscience, Dr. SN Medical College, Jodhpur, India-342001
6. Department of Medicine, Dr. SN Medical College, Jodhpur, India-342001
Received 2017-8-21; Accepted 2017-11-12; Published 2018-4-10
Abstract

Rarity in reporting whole genome sequence of Dengue virus from dengue endemic countries leaves lacunae in understanding regional pattern of virus mutation and ultimately leading to non-understanding of transmission pattern and clinical outcomes emerging at regional levels. Due to inter-serotype genomic similarity and intra-serotype genomic diversity, appropriate designing of primer pairs appears as an exhaustive exercise. Present paper reports new Dengue virus type-specific primer which may help in characterizing virus specific to Asian origin. Genomes of dengue virus serotypes of Asian region were searched and using advanced bioinformatics tools, serotype specific primers were designed and tested for their targeted amplification efficiency. 19 primers sets for DENV-1, 18 primer sets for DENV-2, 17 for DENV-3 and 18 for DENV-4 were designed. In-silico and experimental testing of the designed primers were performed on virus isolated from both clinical isolates and passaged cultures. While all 17 and 18 primer sets of DENV-3 and DENV-2 respectively yielded good quality sequencing results; in case of DENV-4, 16 out of 18 primer sets and in DENV-1, 16 out of 19 primer sets yielded good results. Average sequencing read length was 382 bases and around 82% nucleotide bases were Phred quality QV20 bases (representing an accuracy of circa one miscall every 100 bases) or higher. Results also highlighted importance of use of primer development algorithm and identified genomic regions which are conservative, yet specific for developing primers to achieve efficiency and specificity during experiments.
Keywords: Flavivirus, Mutation, Oligonucleotide, BLAST algorithm, Genetic variation
1. Introduction
Dengue Fever (DF) associated with Dengue Hemorrhagic Fever (DHF) and Dengue Shock Syndrome (DSS) accounts for about 300 million cases reported annually all across the world [1]. In 2016, India observed around 1,11,880 cases and 227 deaths as per data published by the National Vector Borne Disease Control Programme [2]. Besides the fact that dengue is emerging as major viral threat in the country, very few genomic sequences, specific to Indian sub-continent, have been reported [3-6]. Dengue virus has been reported as one of the fastest mutating viruses [7,8] hence to address the increasing disease burden every year, a periodic knowledge of its whole genome sequence is necessary in order to update regional transmission pattern and clinical complexities.
For successful whole genome sequencing of any dengue virus serotype using sanger based method, availability of highly specific and sensitive primers covering whole genome is a critical determinant, as there exists spatiotemporal genomic diversity in individual serotypes leading to auxiliary categorization in lineages and genotypes [9-15]. Also, evidences of concurrent infections of multiple dengue virus serotypes in a single host adds further to the complexity as there is 60 to 75% similarity level in dengue virus serotypes at amino acid level [16-20]. During designing and selection of primers these complexities needs to be over ruled. Considering the challenges of an appropriate primer design for PCR based sequencing of whole genome of dengue virus subtypes, many of the PCR-based whole genome sequencing methods, as reported in the literature, may not function efficiently for every study.
Present paper reports, a simplified yet highly accurate Sanger based sequencing protocol of dengue virus subtypes which has broader applicability. To accomplish this, various bioinformatics tools and programmes were applied to utilize available dengue virus genome sequences data with high level of filtering and optimization. A nearly conservative primer pairs for each dengue virus subtype covering whole genome has been developed with ideal properties and high level of target region specificity and no cross-reactivity to other subtypes.
2. Material and Methods
2.1. Primer Designing
A methodology was employed for identifying genomic regions which were not repetitive and which showed high degree of evolutionary conservation in dengue virus subtype genome. The same design algorithm was employed for primer designing for each serotype. Also, it was ensured that developed primer sets have ideal characteristics of primer pairs, covering amplicon size of 500 - 1000 nucleotide bases and have no cross reactivity to the other than target region of same subtype genome and also no cross reactivity to other subtype genomes. Steps were taken to ensure primers do not have target in host genomes. To accomplish this, available genome sequencing data of each dengue virus serotype, various bioinformatics tools, computer programs and primer design tools along with several well defined filtering steps were used. The approach was iterative so that best primer pairs, for whole genome sequencing were designed. The process of primer design flow is described as flowchart in Figure 1.
Primer design process flow chart
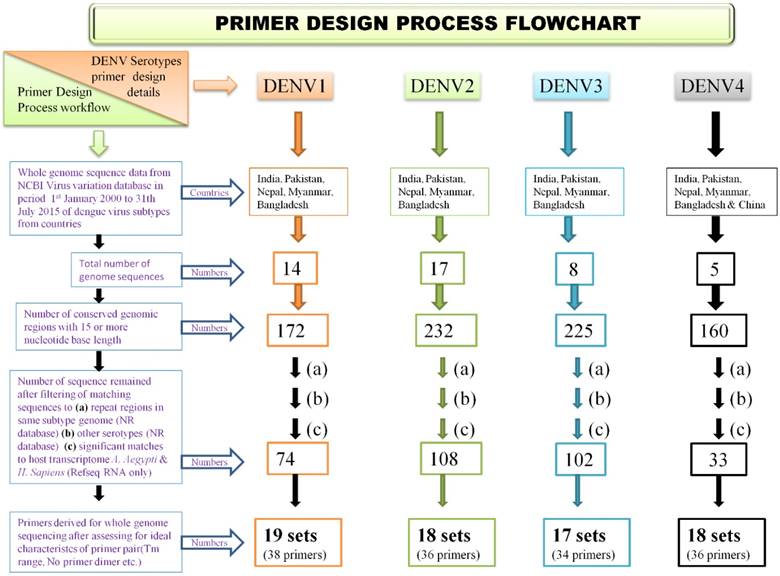
2.2. Genome Selection
For selecting evolutionary conservative genomic regions for primer design multiple sequence alignment (MSA) of available whole genome sequences of a dengue subtype was used. Selection of genomes was a key step as selective filters were required to minimize effect of geographical and time scale variations in dengue viruses. Inclusion of too many genomes would have resulted in very few conservative regions, hence to optimize this, geographical and time scale filters were applied. Whole genomes of a dengue virus subtype reported from countries India, Pakistan, Nepal, Bangladesh and Myanmar (from China also in case of DENV4) isolated in period from January year 2000 to July 2015, were downloaded from NCBI virus variation database [21]. By applying these filters, a total of 14, 17, 8, and 5 complete genomes available for DENV-1, DENV-2, DENV-3 & DENV-4 respectively, were identified whose GenBank accession numbers are provided in the Supplementary file (Table S1).
2.3. Screening of Conserved regions
For each serotype, pairwise local Multiple Sequence Alignment (MSA) was performed using multithreaded program MAFFT (v 7.221) option L-GSSi of above selected genomes [22]. Obtained MSA output was then analyzed in MEGA tool (version 6.0) for identification of conserved genomic sites (constant and singleton sites) [23]. Conserved regions of length 15 bases or more were screened using programs written in Perl programming language. Here we define conserved region a continuous array of conserved genomic sites only.
2.4. Serotype specificity of conserved regions
The serotype specificity of obtained conserved regions was checked by using Basic Local Alignment Search Tool (BLAST). Search method 'megaBLAST' was opted against Non-Reundant (NR) database of the remaining DENV serotypes [24]. Sequences which showed hits (more than five) to other serotypes in BLAST output were not taken further in primer designing process. Sequences which showed no or less than five hits were then taken for primer design.
2.5. Designing Primers from Short sequences
Selection and robust optimization process was performed for these short sequences to obtain desired primer pair candidates. It was ensured that only those primer pairs were selected which have 35% or more GC content, GC lock at 3 prime end, absence of self-annealing or primer dimer possibility with the pair, absence of secondary priming sites, and Melting temperature in a range of 62 to 67˚C. It was also ensured that 3' region of primer is completely from selected conserved sequence so that it remains a highly conserved region. The expected melting temperature (Tm) was calculated using MWG operon primer design tool and care was taken that difference of Tm of forward and reverse primer do not exceed by 5˚Celsius. Also, it was ensured that the amplicon length using primer pair remains within limits of 800-1200 nucleotides. Primer pairs were rejected on the basis of possible primer-dimer formation if a pair had five or more consecutive complementary nucleotides. To ensure complete coverage and end-to-end inclusion, primers for terminal regions were identified to those derived from reference genome with ideal characteristics of good primers but no checks for specificity and complementing them with a highly specific primer so that it forms an specific primer pair. For the purpose of genome positioning and for terminal region primer selection, top most hit of multiple alignment was selected for dengue virus serotypes DENV1 to DENV4 (GenBank accession numbers JN903578, JX475906, GQ466079 & JQ922560 respectively; listed in Supplementary Table S2). The designed primers were then commercially manufactured by m/s Eurofins Genomics Ltd, Bengaluru, India.
2.6. Empirical testing of designed primers employing Reverse Transcriptase- polymerase Chain Reaction (RT-PCR) assay
Sanger Sequencing of DENV serotypes was done using designed primers. In one of the earlier studies [25], whole genome sequencing of DENV-3 isolated from direct clinical serum sample and its C6/36 cell line passage culture sample was performed and reported (GenBank Accession numbers KU216209 & KU216208). The whole genome primers designed for DENV-3 were verified in that study; however sequence and method of primers designed is being reported here. Since no clinical sample positive for DENV-1, 2 and 4, was available during the study period, hence RNA extracted from DENV1-4 positive control virus mix supplied in Dengue virus type detection kit (m/s CDC, Atlanta, USA) was used. The mix contained heat‐inactivated DENV‐1 Haw (Hawaii), DENV‐2 NGC (New Guinea C), DENV‐3 H87, and DENV‐4 H241 strains for which whole genome sequence data was already available in NCBI database (GenBank accession numbers: DENV-1 Haw: EU848545, DENV2 - NGC: AF038403, DENV3 - H87: M93130 and DENV4 - H241: AY947539). The mapping of the primers designed with these corresponding DENV strains referred above is shown in Supplementary table (S3). Using of CDC-positive control virus mix also gave an advantage of protocol applicability if ever concurrent infections by multiple serotypes was to be reported.
RNA was extracted using the QIAamp Viral RNA kit (m/s Qiagen, CA) followed by RT-PCR Step using the One Step superscript -III RT-PCR kit with platinum Taq polymerase (m/s ABI, USA) according to the manufacturer's protocol. For testing each of the sets of designed primers; 19 fragments sets of DENV -1, 18 fragments sets of DENV - 2 and 18 fragment sets of DEN -4 (Total 55 sets; including both forward and reverse primers), a working solution of 1µl (10pmol) was used. The thermal cycling conditions was as follows: Reverse transcription at 55 ° C for 40 minutes, Denaturation at 94° C for 3 minutes, further denaturation of 35 cycles at 94° C for 30 seconds, 50° C for 1 minutes and 68 ° C for 1 minutes and a final extension at 68 ° C for 5 minutes. After amplification, 5µl of each amplicons were run in 2% Agarose gel and bands were obtained. The expected Molecular weight (MW) for each amplified product was approximately 800-1000 nucelotide bases while those amplified with primer sets covering the start and stop points had MW of approximately 450-550 bases (Figure 2).
Dengue virus genome bands obtained as fragments on a 2% Agarose gel. (A) DENV-1 genome bands amplified using 19 sets of primers displayed numerically from lane L1 through L19. (B).DENV-2 genome bands amplified using 18 sets of primers displayed numerically from lane L1 through L18. (c). DENV-4 genome bands amplified using 18 sets of primers displayed numerically from lane L1 through L18.
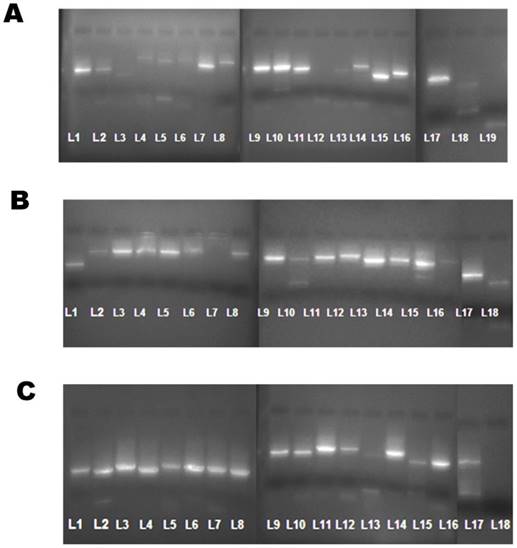
2.7. Amplicon Sequencing
The total 55 amplicons were then subjected to purification using PCR purification kit (QIA quick PCR purification kit, Qiagen, CA). 10µl of purified cDNA's were eluted and subjected to cycle sequencing protocol (Big Dye Terminator v3.1 Ready Reaction -24 mix kit; m/s ABI, USA) using 1 µl of forward primer. Finally to remove any unincorporated dNTP's, the cycle sequenced products were subjected to Dye Ex 2.0 spin kit (m/s Qiagen, CA). These were then subjected to 3130xl Genetic Analyzer (m/s ABI, USA) for obtaining their sequence information.
2.8. Contig assembly
SeqScape v2.6 (Applied Biosystems, USA) was used for quality analysis and optimization of sequenced fragments, contig assembly, reference based mapping of fragments, analysis of variations at nucleotide and amino acid level. NCBI Reference genomes for dengue virus subtypes were used for reference based assembly (DENV1 to DENV4: NC_001477, NC_001474, NC_001475 & NC002640 respectively).
2.9. Integrative analysis
Integrative analysis of primer nucleotide sequences, published genome sequence of DENV serotypes present in positive control virus mix, RT-PCR reaction outcome, sequencing data quality were performed to understand effect of nucleotide mismatch of primer sequence with the published genome of sample whose sequencing is done. Also, to estimate threshold of nucleotide mismatch in total and in the 3 prime end till which good quality sequencing results are obtained. Derived mismatch threshold values can be utilized in insilico testing of primers. Mapping of designed primers to available genome sequence of dengue virus strains present in positive dengue virus control mix was performed using NCBI - BLAST with relaxed search algorithm. Number of variations in nucleotide bases of primers compared to available genome was computed. Nucleotide variations of a pair of primer compared to published sequence were computed and then mapped with RT-PCR outcome and also sequencing quality of reads. Similar analysis were performed for forward primer only of pair, 3 prime end of primer pairs and 3 prime end only of forward primer.
2.10. In-silico testing of primers using e-PCR
For In-silico testing of designed primer pairs, e-PCR tool standalone version was employed {Ref e-PCR}. For all primer pairs of each serotype which were given as query set, two separate searches were performed to two different genome data sets. First set had all genome sequences of that serotype for which primers have to be tested, submitted till 31st July 2015 and isolated in any of South Asian countries (Countries: Afghanistan, Bangladesh, Bhutan, India, Maldives, Nepal, Pakistan and Sri Lanka). In second set, whole Asian countries was taken as sample isolation source with submission time same i.e. 31st July 2015. Decision of geographical region filters was based on fact that the design constraints for primers were so that it has high probability of returning good quality sequencing results of dengue virus strains isolated from Asian or south Asian region. Nucleotide mismatch threshold level derived from integrative analysis were taken as input parameter for e-PCR program.
3. Results
3.1. Primer pairs designing and testing
Through exhaustive computational analysis, a total of 144 primers (72 forward and 72 reverse) were designed covering the four serotypes (listed in suppl table S2).
Of this, 55 primer pairs were used in amplification of DENV 1,2 and 4 and 50 reactions gave results (16 of DENV1, 18 of DENV2 and 16 of DENV4) (Figure 2). Average read length was 386 nucleotide bases and 82% bases were of high quality (Phred score > 20). No sequencing results were obtained for primers 12th, 18th and 19th of DENV1 and 18th pair of DENV4.
The 17 primer pairs used in whole genome sequencing of DENV 3 isolated from patient serum and from passaged culture (as performed in earlier study)[25], showed average read length of 544 bases for serum sample and 560 bases for passaged sample. 87% of nucleotide bases were of high quality (Phred score >20) for clinical isolate while 79% of total bases were of high quality for cell culture passage. Genome coverage of both was the same valued to 1.6x.
3.2. Integrative analysis results
Integrative analysis results suggested possible explanations for negative results to an extent. 18th primer pair for DENV1 had 13 nucleotide mismatches with published sequence which includes 3 mismatches in 3 prime end. Reverse primer of 18th pair for DENV4 had no matching sequence to published genome sequence as published sequence was shorter at 3 prime end compared to from which primer was designed. There were good quality results for either primer of a pair having upto 4 mismatches, primer pair having 8 mismatches, 3 prime end of a either primer of a pair having 1 mismatch and 3 prime end of primer pair combined having 2 mismatch to mapping region of published genome. These mismatch values were then considered threshold levels to be taken up as input for insilico e-PCR based testing (Supplementary file; Table S3).
3.3. e-PCR testing results
Amplification was predicted for all whole genomes as of July 2015 of both South Asian and Asian regions using e-PCR for designed primers. The analysis was performed to predict amplification by designed primers for both set of genomes for all serotypes. Output of reverse e-PCR was then further analysed using Perl programs. Genome sequences for which 15 or more primer pairs predicted amplification were then looked. Threshold 15 pairs was taken to avoid exclusion of genome sequences due to non-mapping of terminal regions primer pairs (Table 1).
Reverse e-PCR results, with designed primers as query set and Genomes set as database.
| Dengue virus subtype | Genomes predicted / Total Genomes (South Asia region, till July 2015) | Genomes predicted / Total Genomes (Asia region, till July 2015) |
|---|---|---|
| DENV1 | 3 /22 | 7/936 |
| DENV2 | 19/23 | 191/483 |
| DENV3 | 31/31 | 276/281 |
| DENV4 | 4 /6 | 15/34 |
4. Discussion and Conclusion
Present paper discusses designing and testing of primer pairs for Sanger based whole genome sequencing of dengue virus serotypes using computational algorithm. These designed primers have ability to amplify reported genomes from Asian and South Asian region, as reported till July, 2015. Instead of designing primers from a single genome, an algorithm to extract relevant sequence information from multiple genomes was taken into consideration. Previous studies also supported that multiple alignment based methods in which identified conserved regions are taken up for designing primers show better efficiency compared to methods in which primers are derived from single gene or genome [25,26]. In addition to identifying conserved regions, the paper also focusses on unique selection of only those conserved regions which showed high degree of dengue virus serotype specificity. The algorithm focuses to minimize effect on sensitivity of primers due to nucleotide variations present in dengue virus subtypes at geographical region and time scale levels. At the same time, it also serves to maximize specificity of primers so as to be efficient in cases of concurrent infections of multiple subtypes.
In case of rapidly evolving organisms such as RNA viruses, the available primers may become ineffective due to risk of insufficient complementation among primers and target sequence. Hence during primer designing it needs to be assured that there remains sufficient complementarity. Magnitude and threshold of complementarity of primers to target sequence so as to get a positive experimental outcome is usually not well defined, herein an attempt to address this by performing an integrative analysis is shown. The analysis suggests that up to eight nucleotide level mismatches in a primer pair with target genome sequence, can also lead to good quality sequencing results. However, nucleotide mismatches more than four and importantly mismatches in 3 prime end region in primers may be a reason for low quality or no sequencing results. These derived values were then given as input parameters in In-silico amplification prediction using e-PCR tool. The prediction results were highly promising and of good applicability to the commonly circulating subtypes with maximum prediction in DENV3 followed by DENV2 subtype. Moderate prediction for DENV4 subtype and least for DENV1 subtype was observed. For pathogens having well identified genotypes or with multiple serotypes, reported methodology has high applicability at individual genotype or serotype level.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Authors are grateful to the Director-General, Indian Council of Medical Research, New Delhi, India for providing financial support to undertake the study and to the Director, Desert Medicine Research Centre, Jodhpur for encouragements and support. The study was supported by the funds provided by Indian Council of Medical Research, India (Biomedical Informatics Center). http://www.icmr.nic.in/. The funders had no role in study design, data collection and analysis and decision to publish or preparation of the manuscript. Authors are also thankful to Center for Disease Control, Atlanta, USA for allowing us to use Positive control virus mix of Dengue virus type detection kit for validation experiments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bhatt S, Gething PW, Brady OJ. et al. The global distribution and burden of dengue. Nature. 2013:504-7
2. http://www.nvbdcp.gov.in/den-cd.html
3. Patil JA, Cherian S, Walimbe AM. et al. Evolutionary dynamics of the American African genotype of dengue type 1 virus in India (1962-2005). Infect Genet Evol. 2011;11:1443-48
4. Cecilia D, Kakade MB, Bhagat AB. et al. Detection of dengue-4 virus in pune, western india after an absence of 30 years-its association with two severe cases. Virol J. 2011;8:46-50
5. Anoop M, Mathew AJ, Jayakumar B. et al. Complete genome sequencing and evolutionary analysis of dengue virus serotype 1 isolates from an outbreak in Kerala, South India. Virus Genes. 2012:1-13
6. Sharma S, Dash PK, Agarwal S. et al. Comparative complete genome analysis of dengue virus type 3 circulating in India between 2003 and 2008. J Gen Virol. 2011;92:1595-1600
7. Drake JW. Rates of spontaneous mutation among RNA viruses. Proc Natl Acad Sci U S A. 1993;90:4171-75
8. Holmes EC, Burch SS. The causes and consequences of genetic variation in dengue virus. Trends Microbiol. 2000;8:74-77
9. Tang Y, Rodpradit P, Chinnawirotpisan P. et al. Comparative analysis of full-length genomic sequences of 10 dengue serotype 1 viruses associated with different genotypes, epidemics, and disease severity isolated in Thailand over 22 years. Am J Trop Med Hyg. 2010;83:1156-65
10. Moya A, Holmes EC, Gonzalez-Candelas F. The population genetics and evolutionary epidemiology of RNA viruses. Nat Rev Microbiol. 2004;2:279-88
11. Rico-Hesse R. Microevolution and virulence of dengue viruses. Adv Virus Res. 2003;59:315-41
12. Repik PM, Dalrymple JM, Brandt WE. et al. RNA fingerprinting as a method for distinguishing dengue 1 virus strains. Am J Trop Med Hyg. 1983;32:577-89
13. Russell PK, McCown JM. Comparison of dengue-2 and dengue-3 virus strains by neutralization tests and identification of a subtype of dengue-3. Am J Trop Med Hyg. 1972;21:97-99
14. Lanciotti RS, Lewis JG, Gubler DJ. et al. Molecular evolution and epidemiology of dengue-3 viruses. J Gen Virol. 1994;75(Pt 1):65-75
15. Henchal EA, Repik PM, McCown JM. et al. Identification of an antigenic and genetic variant of dengue-4 virus from the Caribbean. Am J Trop Med Hyg. 1986;35:393-400
16. Guzman MG, Halstead SB, Artsob H. et al. Dengue: a continuing global threat. Nat Rev Microbiol. 2010;8:S7-16
17. Sim S, Hibberd ML. Genomic approaches for understanding dengue: insights from the virus, vector, and host. Genome Biol. 2016;17:38
18. Wang W-K, Chao D-Y, Lin S-R. et al. Concurrent infections by two dengue virus serotypes among dengue patients in Taiwan. J Microbiol Immunol Infect. 2003;36:89-95
19. Thavara U, Siriyasatien P, Tawatsin A. et al. Double infection of heteroserotypes of dengue viruses in field populations of Aedes aegypti and Aedes albopictus (Diptera: Culicidae) and serological features of dengue viruses found in patients in southern Thailand. Southeast Asian J Trop Med Public Health. 2006;37:468-76
20. Angel B, Angel A, Joshi V. Multiple dengue virus types harbored by individual mosquitoes. Acta Trop. 2015;150:107-10
21. Brister JR, Bao Y, Zhdanov SA. et al. Virus Variation Resource-recent updates and future directions. Nucleic Acids Res. 2014;42:D660-5
22. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772-780
23. Tamura K, Stecher G, Peterson D. et al. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725-2729
24. Altschul SF, Gish W, Miller W. et al. Basic local alignment search tool. J Mol Biol. 1990;215:403-410
25. Angel A, Angel B, Joshi AP. et al. First study of complete genome of Dengue-3 virus from Rajasthan, India: genomic characterization, amino acid variations and phylogenetic analysis. Virology Reports. 2016;6:32-40
26. Lee HK, Tang JW-T, Kong DH-L. et al. Simplified large-scale Sanger genome sequencing for influenza A/H3N2 virus. PLoS One. 2013;8:e64785
27. Gijavanekar C, Anez-Lingerfelt M, Feng C. et al. PCR detection of nearly any dengue virus strain using a highly sensitive primer 'cocktail'. FEBS J. 2011;278:1676-87
Author contact
![]() Corresponding author: Dr. Vinod Joshi, Present Address: Amity Institute of Virology & Immunology (AIVI), Amity University, Noida, U.P., India- 201313. vjoshiedu; 09660767977
Corresponding author: Dr. Vinod Joshi, Present Address: Amity Institute of Virology & Immunology (AIVI), Amity University, Noida, U.P., India- 201313. vjoshiedu; 09660767977