ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2018; 6:30-33. doi:10.7150/jgen.24935 This volume Cite
Short Research Paper
Complete Genome Sequence of Microcystis aeruginosa NIES-2481 and Common Genomic Features of Group G M. aeruginosa
Haruyo Yamaguchi1 ![]() , Shigekatsu Suzuki1, Yasunori Osana2, Masanobu Kawachi1
, Shigekatsu Suzuki1, Yasunori Osana2, Masanobu Kawachi1
1. Center for Environmental Biology and Ecosystem Studies, National Institute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibaraki 305-8506, Japan
2. Department of Electrical and Electronics Engineering, University of the Ryukyus, 1 Senbaru, Nishihara-cho, Okinawa 903-0213, Japan
Received 2018-1-14; Accepted 2018-2-21; Published 2018-3-19
Abstract

Microcystis aeruginosa is a freshwater bloom-forming cyanobacterium that is distributed worldwide. M. aeruginosa can be divided into at least 8 phylogenetic groups (A-G and X) at the intraspecific level. Here, we report the complete genome sequence of M. aeruginosa NIES-2481, which was isolated from Lake Kasumigaura, Japan, and is assigned to group G. The complete genome sequence of M. aeruginosa NIES-2481 comprises a 4.29-Mbp circular chromosome and a 147,539-bp plasmid; the circular chromosome and the plasmid contain 4,332 and 167 protein-coding genes, respectively. Comparative analysis with the complete genome of M. aeruginosa NIES-2549, which belongs to the same group with NIES-2481, showed that the genome size is the smallest level in previously sequenced M. aeruginosa strains, and the genomes do not contain a microcystin biosynthetic gene cluster in common. Synteny analysis revealed only small-scale rearrangements between the two genomes.
Keywords: algal bloom, cyanobacteria, genome, Microcystis
Introduction
Microcystis aeruginosa is a unicellular, colony-forming cyanobacterium distributed in eutrophic freshwater environments worldwide [1]. Blooms of Microcystis during the summer cause serious environmental problems, such as the release of foul odors, bottom layer anoxia, and the production of hepatotoxic cyanotoxins called microcystins. Tanabe et al. first reported a novel method to genetically distinguish M. aeruginosa isolates by using seven housekeeping genes [2], and since then 8 groups (A-G and X) at the intraspecific level have been reported, with the strains in groups A and X, and some strains in group B, being reported to produce microcystins [3]. Tanabe and Watanabe applied the method to many M. aeruginosa strains, and revealed strains assigned to group G mainly distributed only in Lake Kasumigaura, Japan [3].
At the time of the present study, the National Center for Biotechnology Information's Genome database (https://www.ncbi.nlm.nih.gov/genome/genomes/820) contained three complete, and more than 20 draft genome sequences of M. aeruginosa [4, 5, 6]. Of the three complete genome sequences, that of M. aeruginosa NIES-2549—another strain also isolated from Lake Kasumigaura, Japan—was the first reported for a group G strain [7]. Compared with the other two complete genome sequences in the database, the genome of M. aeruginosa NIES-2549 was the smallest (4.3 vs. 5.8 and 5.1 Mbp).
Here, we report the complete genome sequence of M. aeruginosa NIES-2481, which was collected from Lake Kasumigaura at the same time as NIES-2549; a previous phylogenetic study indicated that both strains belong to group G but are genetically distinct [3]. We also report the results of a comparative genomic analysis of the complete genome sequences of M. aeruginosa reported to date, including a synteny analysis of the sequences reported for the two group G strains.
Material and Methods
An axenic culture of M. aeruginosa NIES-2481 was obtained from the Microbial Culture Collection at the National Institute for Environmental Studies, Japan (http://mcc.nies.go.jp/). DNA extraction was performed on a 50-mL culture of NIES-2481 by using NucleoBond Buffer Set III and NucleoBond AXG 500 (Macherey-Nagel) in accordance with the manufacturer's instructions. DNA sequencing was performed by using a PacBio RS II sequencer (Pacific Biosciences). DNA fragmentation was achieved by using a g-TUBE (Covaris); a 20-kb fragment library was constructed, and this was followed by size selection using a BluePippin electrophoresis unit (Sage Science) with the size cut-off set at 15 kb. A single library was prepared and then sequenced in two single-molecule real-time cells by using P6 DNA polymerase and C4 chemistry, yielding a total of 59,021 reads (641,700,882 bp). De novo assembly was performed by means of the Hierarchical Genome Assembly Process [8]. The genome was annotated by using the RAST server [9] or NCBI's Prokaryotic Genome Annotation Pipeline (PGAP) [10]. A chromosome map was drawn by using DNAPlotter [11]. Secondary metabolites were predicted by using antiSMASH [12]. Clustered regularly interspaced short palindromic repeat (CRISPR) loci were detected by using CRISPRFinder [13]. Functional annotation based on cluster of orthologous groups (COG) categories was conducted by using COGNIZER [14]. Synteny analysis was performed by using MURASAKI [15].
Results and Discussion
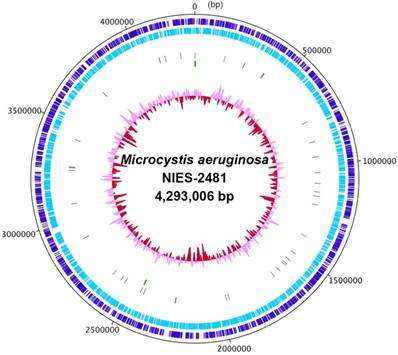
We first determined the complete genome sequence of Microcystis aeruginosa NIES-2481 (Table 1). The genome consists of a 4.29-Mbp circular chromosome (Figure 1) and a 147,539-bp plasmid (read coverages: 114 and 83; G+C contents: 42.91% and 41.66%; protein-coding genes: 4,332 and 167, respectively). The circular chromosome has two sets of rRNA operons and 41 tRNA genes. antiSMASH predicted the presence of 28 secondary metabolite gene clusters, including microviridin [16], aeruginosin [17], and micropeptin biosynthetic gene clusters [18], but not a microcystin biosynthetic gene cluster [19]. CRISPRFinder predicted the presence of five confirmed CRISPR loci in the genome.
General genomic information of Microcystis aeruginosa NIES-2481.
| Features | Chromosome | Plasmid |
|---|---|---|
| Length (bp) | 4,293,006 | 147,539 |
| G+C content (%) | 42.91 | 41.66 |
| rRNA operon | 2 | 0 |
| tRNA genes | 41 | 0 |
| CDS | 4332 | 167 |
Complete chromosome map of Microcystis aeruginosa NIES-2481. The chromosome map comprises five circles. The dark-blue and light-blue circles show the positions of protein-coding genes on the plus and minus strands. The gray bars on the third circle represent tRNA genes. The green bars on the fourth circle represent rRNA genes. The pink/red circle shows the guanine-cytosine content.
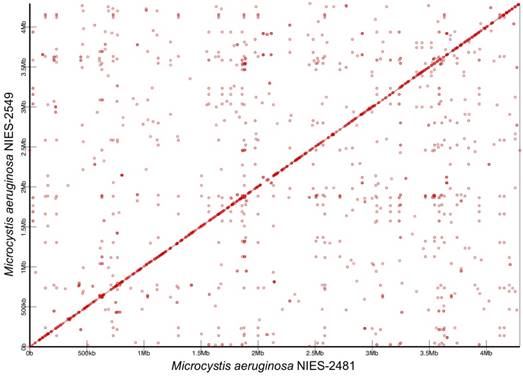
We compared the complete genome of M. aeruginosa NIES-2481 with that of M. aeruginosa NIES-2549, which is the only other group G strain for which a complete genome sequence has been reported [7]. The two genomes are similar in size, synteny, and numbers and kinds of genes. The genome size of NIES-2481 is only 1,207-bp larger than that of NIES-2549. In both strains, the cell harbors one type of plasmid. The genome of NIES-2481 contains slightly more protein-coding genes than does the genome of NIES-2549 (4,332 vs. 4,282). The numbers of rRNA genes and tRNA genes in the two genomes are comparable. The 16S rRNA gene sequences in the two genomes are a 100% match. Functional annotation based on COG categories revealed that the two genomes are well conserved (Table 2) but contain small-scale genome rearrangements (Figure 2).
Comparison of cluster of orthologous groups categories between Microcystis aeruginosa NIES-2481 and NIES-2549.
| category | definition | NIES-2481 | NIES-2549 |
|---|---|---|---|
| B | Chromatin structure and dynamics | 2 | 2 |
| C | Energy production and conversion | 272 | 274 |
| D | Cell cycle control, cell division, chromosome partitioning | 56 | 56 |
| E | Amino acid transport and metabolism | 369 | 371 |
| F | Nucleotide transport and metabolism | 90 | 91 |
| G | Carbohydrate transport and metabolism | 213 | 212 |
| H | Coenzyme transport and metabolism | 210 | 212 |
| I | Lipid transport and metabolism | 105 | 103 |
| J | Translation, ribosomal structure and biogenesis | 227 | 229 |
| K | Transcription | 172 | 172 |
| L | Replication, recombination and repair | 449 | 465 |
| M | Cell wall/membrane/envelope biogenesis | 328 | 330 |
| N | Cell motility | 40 | 41 |
| O | Posttranslational modification, protein turnover, chaperones | 229 | 232 |
| P | Inorganic ion transport and metabolism | 271 | 277 |
| Q | Secondary metabolites biosynthesis, transport and catabolism | 195 | 189 |
| R | General function prediction only | 695 | 727 |
| S | Function unknown | 475 | 467 |
| T | defense mechanisms | 227 | 226 |
| U | Intracellular trafficking, secretion, and vesicular transport | 66 | 66 |
| V | Defense mechanisms | 157 | 155 |
| Z | Cytoskeleton | 1 | 1 |
Synteny analysis of Microcystis aeruginosa NIES-2481 and NIES-2549. Similar genomic regions in the two genomes are indicated by red circles and lines. The horizontal axis represents the genome sequence of M. aeruginosa NIES-2481and the vertical axis represents the genome sequence of M. aeruginosa NIES-2549.
At the time of the present study, aside from the complete genome sequence for M. aeruginosa NIES-2549, the only other complete M. aeruginosa genome sequences that have been reported are for strains NIES-843 (5.8 Mbp) [4] and PCC7806SL (5.1 Mbp) (https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP020771.1); M. aeruginosa NIES-843 is assigned to group A [3], but M. aeruginosa PCC7806SL is yet to be assigned to a group. Compared with the genomes of strains NIES-2481 and NIES-2549, those of strains NIES-843 and PCC7806SL are approximately 1 Mbp larger. This difference in size is explained by the different numbers of genes contained by the genomes. The genomes of strains NIES-843 and PCC7806SL contain a microcystin biosynthetic gene cluster, whereas those of strains NIES-2481 and NIES-2549 do not. However, the microcystin biosynthetic gene cluster is only approximately 55 kbp, and therefore the smaller genome size of the clade G strains is not fully explained by the absence of this gene cluster. Functional annotation based on COG categories indicated that a major difference between the genomes of strains NIES-843 and NIES-2481 is the number of category L (replication, recombination, and repair) genes (1,037 vs. 449 genes, respectively). Especially, these differences are caused by number of transposases, and these would mainly affect gaps of genome size between the two genomes. A comparative genome analysis of M. aeruginosa by Humbert et al. has suggested that the M. aeruginosa genome encodes a high proportion of transposases, and those can permit rapid variation in the genome and survive harsh freshwater environments [6]. However, the group G strains of M. aeruginosa possess a smaller genome than the other reported genomes, and their genomes contain fewer transposase-encoding genes than that of a strain in group A. The small genome size may facilitate rapid DNA replication and cell growth. That is, the relatively small genome size may provide the strains in this group with a growth advantage over other strains of M. aeruginosa. Tanabe and Watanabe reported that the cell size of strains in group G is larger than that of strains in other groups and that the cells aggregate loosely to form small, irregular colonies [3]. Limnological reasons for dominancy of group G in the lake are unknown, however, these differences in genome size and morphological features imply that group G strains of M. aeruginosa have specifically evolved to adapt to the recent environment in Lake Kasumigaura. To further understand the genomic evolution and ecology of group G strains of M. aeruginosa, additional complete genome analyses of strains in all of the M. aeruginosa groups are required.
Accession Numbers
This Whole Genome Shotgun project of M. aeruginosa NIES-2481 has been deposited in NCBI under the accession no. CP012375 (chromosome) and CP025929 (plasmid).
Acknowledgements
This study was supported by the Japan Agency for Medical Research and Development (AMED).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Watanabe MF, Harada KI, Carmichael WW, Fujiki H editors. Toxic Microcystis. CRC Press, Boca Raton, FL. 1996
2. Tanabe Y, Kasai F, Watanabe MM. Multilocus sequence typing (MLST) reveals high genetic diversity and clonal population structure of the toxic cyanobacterium Microcystis aeruginosa. Microbiology. 2007;153:3695-3703
3. Tanabe Y, Watanabe MM. Local expansion of a panmictic lineage of water bloom-forming cyanobacterium Microcystis aeruginosa. PLoS One. 2011;6:e17085
4. Kaneko T, Nakajima N, Okamoto S. et al. Complete genomic structure of the bloom-forming toxic cyanobacterium Microcystis aeruginosa NIES-843. DNA Res. 2007;14:247-256
5. Frangeul L, Quillardet P, Castets AM. et al. Highly plastic genome of Microcystis aeruginosa PCC 7806, a ubiquitous toxic freshwater cyanobacterium. BMC Genomics. 2008;9:274
6. Humbert JF, Barbe V, Latifi A. et al. A tribute to disorder in the genome of the bloom-forming freshwater cyanobacterium Microcystis aeruginosa. PLoS One. 2013;8:e70747
7. Yamaguchi H, Suzuki S, Tanabe Y. et al. Complete genome sequence of Microcystis aeruginosa NIES-2549, a bloom-forming cyanobacterium from Lake Kasumigaura, Japan. Genome Announc. 2015;3:e00551-15
8. Chin CS, Alexander DH, Marks P. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat methods. 2013;10:563-569
9. Aziz RK, Bartels D, Best AA. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75
10. Tatusova T, DiCuccio M, Badretdin A. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614-6624
11. Carver T, Thomson N, Bleasby A. et al. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2008;25:119-120
12. Weber T, Blin K, Duddela S. et al. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237-W243
13. Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35(Suppl 2):W52-W57
14. Bose T, Haque MM, Reddy CVSK. et al. COGNIZER: a framework for functional annotation of metagenomic datasets. PLoS One. 2015;10:e0142102
15. Popendorf K, Tsuyoshi H, Osana Y. et al. Murasaki: a fast, parallelizable algorithm to find anchors from multiple genomes. PLoS One. 2010;5:e12651
16. Ziemert N, Ishida K, Weiz A. et al. Exploiting the natural diversity of microviridin gene clusters for discovery of novel tricyclic depsipeptides. Appl Environ Microbial. 2010;76:3568-3574
17. Ishida K, Welker M, Christiansen G. et al. Plasticity and evolution of aeruginosin biosynthesis in cyanobacteria. Appl Environ Microbial. 2009;75:2017-2026
18. Nishizawa T, Ueda A, Nakano T. et al. Characterization of the locus of genes encoding enzymes producing heptadepsipeptide micropeptin in the unicellular cyanobacterium Microcystis. J Biochem. 2011;149:475-485
19. Tillett D, Dittmann E, Erhard M. et al. Structural organization of microcystin biosynthesis in Microcystis aeruginosa PCC7806: an integrated peptide-polyketide synthetase system. Chem Biol. 2000;7:753-764
Author contact
![]() Corresponding author: Haruyo Yamaguchi, Center for Environmental Biology and Ecosystem Studies, National Institute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibaraki 305-8506, Japan. Tel: +81-29-850-2424. Fax: +81-29-850-2587. E-mail: yamaguchi.haruyogo.jp.
Corresponding author: Haruyo Yamaguchi, Center for Environmental Biology and Ecosystem Studies, National Institute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibaraki 305-8506, Japan. Tel: +81-29-850-2424. Fax: +81-29-850-2587. E-mail: yamaguchi.haruyogo.jp.