ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2017; 5:58-63. doi:10.7150/jgen.20358 This volume Cite
Research Paper
CNV Analysis of Host Responses to Porcine Reproductive and Respiratory Syndrome Virus Infection
El Hamidi A. Hay1*, Igseo Choi2*, Lingyang Xu1, 3, Yang Zhou1, 4, Robert R. R. Rowland5, Joan K. Lunney2 ![]() , George E. Liu1
, George E. Liu1 ![]()
1. Animal Genomics and Improvement Laboratory, BARC, USDA-ARS, Beltsville, Maryland 20705, USA;
2. Animal Parasitic Diseases Laboratory, BARC East, USDA-ARS, Beltsville, Maryland 20705, USA.
3. Institute of Animal Science, Chinese Academy of Agricultural Science, Beijing, 100193, China;
4. College of Animal Science and Technology, Northwest A&F University, Shaanxi Key Laboratory of Agricultural Molecular Biology, Yangling, Shaanxi 712100, China.
5. Department of Diagnostic Medicine and Pathobiology, College of Veterinary Medicine, Kansas State University, Manhattan, KS 66506, USA.
* Co- first authors.
Published 2017-5-24
Abstract

Porcine reproductive and respiratory syndrome (PRRS) is a devastating disease with a significant impact on the swine industry causing major economic losses. The objective of this study is to examine copy number variations (CNVs) associated with the group-specific host responses to PRRS virus infection. We performed a genome-wide CNV analysis using 660 animals genotyped with on the porcine SNP60 BeadChip and discovered 7097 CNVs and 271 CNV regions (CNVRs). For this study, we used two established traits related to host response to the virus, i.e. viral load (VL, area under the curve of log-transformed serum viremia from 0 to 21 days post infection) and weight gain (WG42 from 0 to 42 days post infection). To investigate the effects of CNVs on differential host responses to PRRS, we compared groups of animals with extreme high and low estimated breeding values (EBVs) for both traits using a case-control study design. For VL, we identified 163 CNVRs (84 Mb) from the high group and 159 CNVRs (76 Mb) from the low group. For WG42, we detected 126 (68 Mb) and 156 (79 Mb) CNVRs for high and low groups, respectively. Based on gene annotation within group-specific CNVRs, we performed network analyses and observed some potential candidate genes. Our results revealed these group-specific genes are involved in regulating innate and acquired immune response pathways. Specifically, molecules like interferons and interleukins are closely related to host responses to PRRS virus infection.
Keywords: PRRS, Copy Number Variation, SNP.
Introduction
Porcine reproductive and respiratory syndrome (PRRS) is the most economically important disease caused by a contagious RNA virus for the porcine industry. Infection with PRRS virus (PRRSV) leads to reproductive losses, slow growth rate, pneumonia and, for certain highly pathogenic isolates, high rates of mortality. Eradicating PRRS or controlling it through vaccination has been difficult due to the structural nature of the virus and its high mutation rate [1, 2]. Another plausible approach to control PRRS is the utilization of genetic variation to select genetically resistant pigs [3]. Multiple studies have reported a genetic component to the resistance to PRRS virus [4]. Using SNP markers, Boddicker et al. found a major QTL on Sus scrofa chromosome (SSC) 4 associated with host response to PRRS virus measured as viral load i.e. VL, the area under the curve of log viremia in blood up to 21 days post-infection (dpi), and weight gain (WG42, gain from 0 to 42 dpi) in growing pigs [5]. Additionally, Boddicker et al. further validated the results and showed that the two traits associated with host response are controlled by multiple regions in the genome with small effects (<1.5%) except the region in SSC4 which explained 13.2 % of the total genetic variation for VL trait [6,7]. Based on differential gene expression in blood samples, Koltes et al. affirmed that alleles in the GBP5 gene account for the SSC4 effect. These findings offer the opportunity to use a marker-assisted type of selection to eliminate or mitigate the impact of PRRS [8].
Utilizing genetic improvement is a good method to control PRRS, however these studies also indicated that SNP markers may not be sufficient to capture all of the genetic variances of the host response to the virus. Therefore, other sources of genetic variation such as copy number variations (CNVs) can possibly contribute to the genetic portion of PRRS traits. CNVs are a subset of structural variations in the forms of insertions and deletions of a size larger than 50bp [9]. Several studies have shown CNVs to alter gene structure, dosage and gene regulation and expose recessive alleles [10]. A human study showed that CNVs explain around 18% of the total variation in gene expression [11]. Additionally, CNVs are of great importance in livestock, having significant effects on economically important traits such as milk production, feed efficiency and disease resistance [12-14]. For the porcine industry, previous CNV studies have produced several CNV maps across the Sus Scrofa genome. Fadista et al. identified 37 CNVRs on chromosomes 4, 7, 14 and 17 and Ramayo-Caldas et al. found 49 CNVRs in Iberian × Landrace crossbred animals using Porcine SNP60 BeadChip [15,16]. Recently, Chen et al. investigated the distribution of 565 CNV regions (CNVRs) from 18 diverse pig populations. However, none of these studies have investigated the correlation between CNVs and complex disease traits in pigs. Thus, to explore the other genetic variations related to PRRS beyond SNPs, the objective of this study is to conduct a group-specific CNV analysis by contrasting CNVRs detected in two groups of extreme traits. Furthermore, network analyses were carried out to understand the functional roles of the detected CNVs on differential host responses to PRRS virus.
Materials and Methods
Data generation
The collection and data generation process were described previously [5]. In total, 660 pigs infected with PRRS virus (NVSL97-7895) were considered in this study. Two traits were established before as indicatives of host response to PRRS. These traits were viral load (VL, area under the curve of log-transformed serum viremia from 0 to 21 days post infection) and weight gain (WG42 from 0 to 42 days post infection) [5].
We calculated Estimated Breeding Values (EBVs) using a single trait model in BLUPF90 package [17]. The model used is the following:
(1)
where y is the vector of observations, b is a vector of fixed factors including sex, pen within trial and the interaction of trial and parity class, a is the vector of random additive effects and e is the vector or residual terms. X and Z are incidence matrices relating effects fixed effects b and random effects a respectively to the observations. Based on the predicted EBVs for VL and WG42, we selected 100 individuals with extremely low values (VLL or WG42L) or 100 individuals with extremely high values (VLH or WG42H) for group comparisons, respectively.
CNV discovery
We detected CNVs using the PennCNV algorithm [18]. PennCNV has the lowest false discovery rate and has been widely used for detecting CNVs compared to other CNV calling algorithms [19]. Multiple sources of information used in PennCNV consist of normalized total intensities (Log R ratio - LRR) and allelic intensity ratios (B allele frequency - BAF). Illumina GenomeStudio software was used to generate both LRR and BAF files. Also, a population frequency of B allele (PFB) file was generated by calculating the BAF of each marker using this population. Additionally, in order to correct for potential waviness due to GC content, genomic_wave.pl option was used and the required gcmodel file was generated by calculating the GC content of the 1Mb genomic region surrounding each marker (500kb each side). In this study like many other published studies, CNV calling was restricted to autosomes only. After sample quality control, we detected 7097 CNVs from 660 animals, which passed the quality control filtering. The quality control filtering parameters were set to 0.35 for standard deviation (STD) of log R ratio, 0.075 for the waviness factor, 0.01 for BAF drift and a threshold of 100 low quality CNVs to eliminate samples (-qclrrsd 0.35 -qcwf 0.075 -qcbafdrift 0.01 -qcnumcnv 100 -qcpassout). After aggregating the overlapping CNVs, we identified a total of 271 CNVRs (Table 1 and Table S1).
Summary of CNV regions of extreme groups
| Sample | Trait | Counta | Gainb | Lossc | Total length |
|---|---|---|---|---|---|
| All samples | VL,WG42 | 271 | 319 | 6778 | 186,923,627 |
| VLL | VL low | 159 | 55 | 911 | 76,462,440 |
| VLH | VL high | 163 | 32 | 969 | 84,558,089 |
| WG42L | WG42 low | 156 | 64 | 1025 | 79,628,510 |
| WG42H | WG42 high | 126 | 41 | 1000 | 68,708,828 |
a Number of non-redundant CNV regions in a specific sample group
b Number of gain events in the identified CNV regions
c Number of loss events in the identified CNV regions
CNV gene annotation and network identification
After the identification of CNV regions for each group, we performed gene annotation using either Ensembl genes (http://www.ensembl.org/) or RefSeq genes. It is noted that these two gene datasets are currently not complete due to the current status of the pig draft genome assembly and annotation. This limitation could have implications on this study.
In order to explore the potential CNVs underlying genetic mechanisms in differential host responses to PRRS virus infection, we carried out network analyses for both group comparisons using Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA, USA). We imported lists of unique genes identified in CNV regions for two group comparisons into IPA separately. We selected "human, mouse and rat" for the species option and included "both in silico and experimental" for Evidence as described previously [20]. We chose the default number of 35 molecules in each network. The networks involving the unique genes in each group were then identified. Furthermore, the genes in the networks which were referred to as focus molecules were classified by their function and assigned a value indicating the significance of the genes in the network.
Results and Discussion
Using the PennCNV algorithm, we identified 7097 CNVs from 660 samples. We obtained a total of 271 CNVRs with a length of 186.9 Mb, which corresponds to approximately ≈ 6% of the pig autosomal genome. The detected 271 CNVRs consisted of 319 gain events and 6778 loss events (Table 1 and Table S1). For the VLH sample, we identified 163 CNV regions with a length of 84 Mb consisting of 32 gain events and 969 loss events (Table S2). Additionally, for the VLL sample, we detected a total of 159 CNV regions (76 Mb) consisting of 55 gain events and 911 loss events (Table S3). We observed a similar trend in the WG42H and WG42L groups (Table S4 and Table S5). Our study revealed that more CNV loss events were identified compared to CNV gain events in all samples, which is consistent with previous studies in pig and other species [21, 22].
CNV correlation with PRRS
In this study, we divided animals into extreme groups based on two traits, thus CNVR for each group was obtained by merging CNVs across individuals into non-overlap regions. To study the potential genes involved in copy number changes, we investigated group-specific gene content using either Ensembl or RefSeq gene datasets. We focused on these group-specific genes because they can shed light on the underlying genetic mechanism involved in differential host responses to PRRS virus infection. As expected, the Ensembl dataset yielded a higher number of genes falling within CNVRs than RefSeq. Using Ensembl, we retrieved 225 genes for the VLH group and 142 genes for the VLL group (Figure 1A). For WG42, we identified 143 and 248 genes for WG42H and WG42L groups, respectively (Figure 1B). When using the RefSeq database, we found 115 genes for the VLH group and 113 genes for the VLL groups. For WG42, we found 101 and 119 genes for WG42H and WG42L groups, respectively. The moderate number of genes found in this study may be related to the location of CNVs in gene-poor regions of the genome and the deleterious nature of CNVs in gene-rich regions [23]. This could be also related to incomplete gene annotation or the sequence quality in the region where a gene is located in the pig genome. For example, we found some CNV regions harboring no annotated gene and these included 35 out of 163 (21%) for the VLH group, 38 out of 159 (23%) for the VLL group, 24 out of 126 (19%) for the WG42H group and 35 out of 156 (22%) for the WG42L group, respectively. Around 20% of CNVR discovered in this study overlapped with previous studies [16, 21, 22, 24]. The discrepancies could be related to different samples and/or CNV calling algorithms [19].
A, Comparison of identified CNV regions between the VLL and VLH groups. B, Comparison of identified CNV regions between WG42H and WG42L groups. C, The top network for the WG42H group identified by IPA is involved in Developmental Disorder, Hereditary Disorder, Immunological Disease. Notes and edges are displayed with various shapes and labels that represent the functional class of genes and the nature of the relationship between the notes, respectively. For meanings of shapes and lines, see legend within the figure.
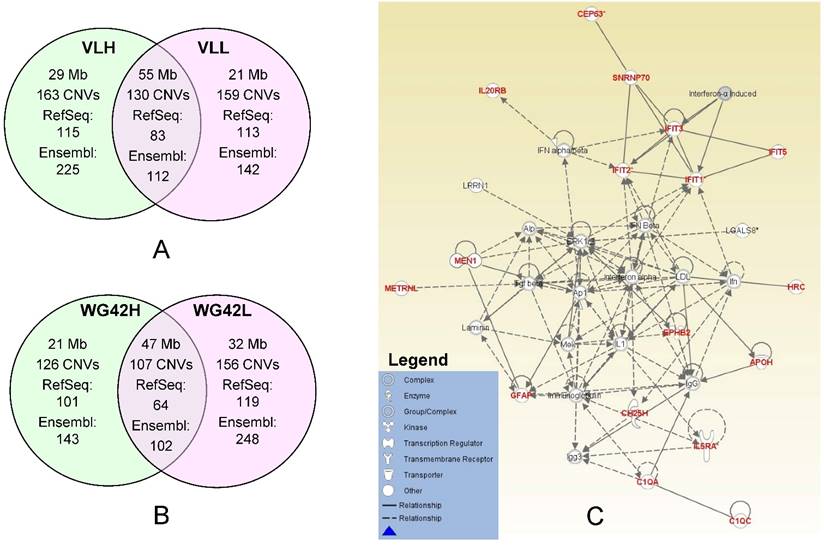
To investigate the effects of CNVs on the host response to PRRS, we carried out network analyses using each set of group-specific genes. Using the IPA software, we obtained various networks for each group. We discovered 8 networks for the VLL group versus 10 networks for the VLH group (Table S6). For WG42, we identified 12 networks for the WG42L group and 7 networks for the WG42H group (Table S7). The score of each network reported by IPA represents the probability that the genes in the network are not associated by random chance.
Interestingly, we discovered a network with gene expression and hereditary functions for the VLH group (Table S6, VLH ID 2). The network contained inflammatory cytokines such as type 1 interferons (IFN-α, IFN-β), TNF-α, and interleukin-1 (IL-1). We also found a network related to antimicrobial response, inflammatory response, cell death and survival for the VLL groups (Table S6 VLL ID 6). The CNV-associated genes found in this network are BFSP2, CALCOCO2, CHMP6, CSGALNACT1, ERICH6, HOXB8, KANSL1, KCNH8, PPM1F, SCN9A, and SLC28A3. The gene CHMP6 encodes a member of the chromatin-modifying protein/charged multivesicular body protein family. These proteins are part of the endosomal sorting complex required for transport III which degrades surface receptors and also part of biosynthesis of endosomes.
Network analysis of the WG42H group resulted in a total of 7 networks. The network with the highest score is involved in developmental disorder, hereditary disorder and immunological disease functions (Figure 1C, Table S7 WG42H ID: 1). The genes in this network included IFIT2, IFIT2, IFIT3 and IFIT5. The IFIT proteins are involved in response processes to viral infections. Infection with PRRS virus activates IFIT1 and IFIT3 expression in porcine alveolar macrophages, and expression of IFIT1 and IFIT5 in the lung [25, 26]. The network also contained inflammatory cytokines such as type 1 interferons (IFN-α, IFN-β), TNF-α, and interleukin-1 (IL-1). These molecules are mainly involved in communication between innate and adaptive immune cells. Earlier studies showed that the PRRS virus under-regulates the production of the aforementioned inflammatory cytokines [27, 28]. Furthermore, their decreased production leads to a weak innate immune response and a slow IFN- γ response [29]. Lunney et al. reported that animals which cleared PRRSV infection were characterized by early expression of IL-1β, IL-8 and IFN-γ [30]. In the WG42L group, we identified a network responsible for humoral immune response, protein synthesis, hematological system development and function (Table S7 WG42L ID: 4). The genes identified in the CNV regions in this network are ANKRD2, CCR7, GALNT2, IK2F3, IL27RA, IRF2, ITPKB, LGI1, PCSK9, RBP4, RLN3, TCAP, TF, TLR5 and TOB1. The pathways in this network also include different interferon and interleukin molecules.
Conclusions
This study revealed that CNVs are potentially involved with group-specific host responses to PRRS virus. Agreeing with previous studies, these results revealed that different interferon and interleukin molecules, which are mainly involved in communication between innate and adaptive immune cells, could be involved in host-PRRS virus interaction and PRRS resistance. This study may benefit the porcine industry by paving the way to utilize genetic variation, specifically CNVs, as a remedy or an approach to eliminate or mitigate the impact of PRRS. Combined with the SNP-based results, our CNV results could potentially facilitate the identification of susceptible animals or allow use of marker assisted type of selection to alleviate the effect of this disease. Finally, it is worthwhile to note that this CNV study is still preliminary in nature and more research is warranted to establish a cause-and-effect relationship in the future.
Supplementary Material
Table S1. CNVRs identified in this study and its state. Table S2. CNVRs identified for the VLH groups. Table S3. CNVRs identified for the VLL samples. Table S4. CNVRs identified for the WG42H group. Table S5. CNVRs identified for the WG42L groups. Table S6. IPA pathways identified for the VL group comparison. Table S7. IPA pathways identified for the WG42 group comparison.
Acknowledgements
We thank the PRRS Host Genetics Consortium for sharing their SNP data. We also thank Reuben Anderson and Alexandre Dimtchev for technical assistance. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture.
Additional Information
This work was supported in part by Agriculture and Food Research Initiative (AFRI) competitive grant No. 2011-67015-30183 from the USDA National Institute of Food and Agriculture (NIFA) Animal Genome Program and AFRI No. 2008-55620-19132 and National Pork Board grants to the PRRS Host Genetics Consortium. The genotypic and phenotypic data was collected as part of the PRRS Host Genetics Consortium (PHGC) studies. All the data is available through the database at http://www.animalgenome.org/lunney/. As noted on that page access to PHGC Database is monitored by the USDA ARS maintained Cooperative Research and Development Agreement (CRADA) Material Transfer Agreement (MTA). Access to this data is available for further analyses to those who sign on to the PHGC CRADA MTA.
Authors' contributions
GEL, IC and EHAH conceived and designed the experiments. EHAH, JL, IC, LYX, YZ and RRRR performed in silico prediction and computational analyses. GEL, EHAH, and JL wrote the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kimman TG, Cornelissen LA, Moormann RJ, Rebel JM, Stockhofe-Zurwieden N. Challenges for porcine reproductive and respiratory syndrome virus (PRRSV) vaccinology. Vaccine. 2009;27:3704-3718
2. Geldhof MF, Van Breedam W, De Jong E, Lopez RA, Karniychuk UU, Vanhee M. et al. Antibody response and maternal immunity upon boosting PRRSV-immune sows with experimental farm-specific and commercial PRRSV vaccines. Vet Microbiol. 2013;167:260-271
3. Rowland RR, Lunney J, Dekkers J. Control of porcine reproductive and respiratory syndrome (PRRS) through genetic improvements in disease resistance and tolerance. Front Genet. 2012;3:260
4. Lunney JK, Chen H. Genetic control of porcine reproductive and respiratory syndrome virus responses. Virus Res. 2010;3:161-169
5. Boddicker N, Waide EH, Rowland RR, Lunney JK, Garrick DJ, Reecy JM. et al. Evidence for a major QTL associated with host response to porcine reproductive and respiratory syndrome virus challenge. J Anim Sci. 2012;90:1733-1746
6. Boddicker NJ, Bjorkquist A, Rowland RR, Lunney JK, Reecy JM, Dekkers JC. Genome-wide association and genomic prediction for host response to porcine reproductive and respiratory syndrome virus infection. Genet Sel Evol. 2014;46:18
7. Boddicker NJ, Garrick DJ, Rowland RR, Lunney JK, Reecy JM, Dekkers JC. Validation and further characterization of a major quantitative trait locus associated with host response to experimental infection with porcine reproductive and respiratory syndrome virus. Anim Genet. 2014;45:48-58
8. Koltes JE, Fritz-Waters E, Eisley CJ, Choi I, Bao H, Kommadath A. et al. Identification of a putative quantitative trait nucleotide in guanylate binding protein 5 for host response to PRRS virus infection. BMC genomics. 2015;16:412
9. Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C. et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59-65
10. Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451-481
11. Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N. et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848-853
12. Glick G, Shirak A, Seroussi E, Zeron Y, Ezra E, Weller JI. et al. Fine Mapping of a QTL for Fertility on BTA7 and Its Association With a CNV in the Israeli Holsteins. G3 (Bethesda ). 2011;1:65-74
13. Kadri NK, Sahana G, Charlier C, Iso-Touru T, Guldbrandtsen B, Karim L. et al. A 660-Kb deletion with antagonistic effects on fertility and milk production segregates at high frequency in Nordic Red cattle: additional evidence for the common occurrence of balancing selection in livestock. PLoS Genet. 2014;10:e1004049
14. Xu L, Cole JB, Bickhart DM, Hou Y, Song J, VanRaden PM. et al. Genome wide CNV analysis reveals additional variants associated with milk production traits in Holsteins. BMC genomics. 2014;15:683
15. Fadista J, Nygaard M, Holm LE, Thomsen B, Bendixen C. A Snapshot of CNVs in the Pig Genome. PloS one. 2008;3:12
16. Ramayo-Caldas Y, Castello A, Pena RN, Alves E, Mercade A, Souza CA. et al. Copy number variation in the porcine genome inferred from a 60 k SNP BeadChip. BMC genomics. 2010;11:593
17. Misztal I, Tsuruta S, Strabel T, Auvray B, Druet T, Lee DH. BLUPF90 and related programs (BGF90). Proceedings of the 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France. 2002;28:07
18. Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF. et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665-1674
19. Winchester L, Yau C, Ragoussis J. Comparing CNV detection methods for SNP arrays. Brief Funct Genomic Proteomic. 2009;8:353-366
20. Hou Y, Bickhart DM, Chung H, Hutchison JL, Norman HD, Connor EE. et al. Analysis of copy number variations in Holstein cows identify potential mechanisms contributing to differences in residual feed intake. Funct Integr Genomics. 2012;12:717-723
21. Wang L, Liu X, Zhang L, Yan H, Luo W, Liang J. et al. Genome-wide copy number variations inferred from SNP genotyping arrays using a Large White and Minzhu intercross population. PLoS ONE. 2013;8:e74879
22. Schiavo G, Dolezal MA, Scotti E, Bertolini F, Calo DG, Galimberti G. et al. Copy number variants in Italian Large White pigs detected using high-density single nucleotide polymorphisms and their association with back fat thickness. Anim Genet. 2014;45:745-749
23. Conrad DF, Hurles ME. The population genetics of structural variation. Nat Genet. 2007;39:S30-S36
24. Wang J, Jiang J, Fu W, Jiang L, Ding X, Liu JF. et al. A genome-wide detection of copy number variations using SNP genotyping arrays in swine. BMC genomics. 2012;13:273
25. Xiao S, Mo D, Wang Q, Jia J, Qin L, Yu X. et al. Aberrant host immune response induced by highly virulent PRRSV identified by digital gene expression tag profiling. BMC genomics. 2010;11:544
26. Zhou P, Zhai S, Zhou X, Lin P, Jiang T, Hu X. et al. Molecular characterization of transcriptome-wide interactions between highly pathogenic porcine reproductive and respiratory syndrome virus and porcine alveolar macrophages in vivo. Int J Biol Sci. 2011;7:947-959
27. Van Reeth K, Labarque G, Nauwynck H, Pensaert M. Differential production of proinflammatory cytokines in the pig lung during different respiratory virus infections: correlations with pathogenicity. Res Vet Sci. 1999;67:47-52
28. Thanawongnuwech R, Young TF, Thacker BJ, Thacker EL. Differential production of proinflammatory cytokines: in vitro PRRSV and Mycoplasma hyopneumoniae co-infection model. Vet Immunol Immunopathol. 2001;79:115-127
29. Costers S, Lefebvre DJ, Goddeeris B, Delputte PL, Nauwynck HJ. Functional impairment of PRRSV-specific peripheral CD3+CD8high cells. Vet Res. 2009;40:46
30. Lunney JK, Fritz ER, Reecy JM, Kuhar D, Prucnal E, Molina R. et al. Interleukin-8, interleukin-1beta, and interferon-gamma levels are linked to PRRS virus clearance. Viral Immunol. 2010;23:127-134
Author contact
![]() Corresponding authors: GEL: Animal Genomics and Improvement Laboratory, USDA-ARS, Building 306, Room 111, BARC-East, Beltsville, MD 20705, USA. E-mail: George.Liuusda.gov, Voice Phone: +1-301-504-9843, Fax: +1-301-504-8414. JKL: Animal Parasitic Diseases Laboratory, BARC East, USDA-ARS, Beltsville, Maryland 20705, USA; E-mail: Joan.Lunneyusda.gov, Voice Phone: +1-301-504-9368.
Corresponding authors: GEL: Animal Genomics and Improvement Laboratory, USDA-ARS, Building 306, Room 111, BARC-East, Beltsville, MD 20705, USA. E-mail: George.Liuusda.gov, Voice Phone: +1-301-504-9843, Fax: +1-301-504-8414. JKL: Animal Parasitic Diseases Laboratory, BARC East, USDA-ARS, Beltsville, Maryland 20705, USA; E-mail: Joan.Lunneyusda.gov, Voice Phone: +1-301-504-9368.