ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2016; 4:13-15. doi:10.7150/jgen.14847 This volume Cite
Short Research Communication
Draft Genome Sequence of Paracoccus sp. MKU1, a New Bacterial Strain Isolated from an Industrial Effluent with Potential for Bioremediation
Kamaldeen Nasrin Nisha1, Jayavel Sridhar2, Perumal Varalakshmi3, Balasubramaniem Ashokkumar1 ![]()
1. Department of Genetic Engineering, School of Biotechnology, Madurai Kamaraj University, Madurai, Tamil Nadu.
2. Department of Biotechnology, Directorate of Distance Education, Madurai Kamaraj University, Madurai, Tamil Nadu.
3. Department of Molecular Microbiology, School of Biotechnology, Madurai Kamaraj University, Madurai, Tamil Nadu.
Published 2016-4-26
Abstract
Paracoccus sp. MKU1, a novel dimethylformamide degrading bacterial strain was originally isolated from an industrial effluent, Tirupur region, Tamil Nadu, India. Here, we report the draft genome sequence of Paracoccus sp. MKU1, which could provide the genetic insights on its evolution and application of this versatile bacterium for effective degradation of xenobiotics and thus in bioremediation.
Keywords: Paracoccus sp. MKU1, genome, dimethylformamide, xenobiotics, degradation
Introduction
Paracoccus is a gram negative coccoid bacterium, which comprises a wide number of species and comes under Rhodobacteraceae family. Paracoccus species exhibit profound genomic divergence and appear to exist as evolutionary continuum. Members of this genera are versatile exhibiting a tremendous range of metabolic flexibility, utilizing a variety of organic compounds including potentially hazardous pollutants (1) such as N,N-dimethylformamide (2), pyridine (3), chloroacetamides (4), N-methylpyrrolidone (5) and known for its adaptation to the prevailing environmental conditions. These physiological characteristics make it as the suitable bacteria for biodegradation of recalcitrant compounds. Many studies on the attributes of multifaceted Paracoccus towards utilization of various compounds are underway and continued to be an emerging area of interest in bioremediation.
Previously, we isolated a dimethylformamide (DMF) degrading Paracoccus sp. MKU1 from the textile industrial effluent near Tirupur, Tamil Nadu, India (6) and the genome of Paracoccus sp. MKU1 has been described herein. The genomic DNA was isolated and the whole genome sequencing was performed using the MiSeq Illumina sequencing platform. Paired end reads with the insert size of 300 bp has achieved 100 fold genome coverage with the Paracoccus sp. MKU1 genome. The raw sequence reads R1 and R2 of about 1,609,204 reads respectively generated by Illumina sequencing were merged using PEAR software tool (7) and the de novo assembly was done using DNASTAR's SeqMan Ngen software (version 12.0). The assembly yielded 242 contigs (N50 contig size, approximately 58 Kb) accounting for a total length of 5.3 Mbp with the largest and smallest contigs of about 338,971 bp and 628 bp respectively. Analysis of Paracoccus sp. MKU1 genome assembly showed a size of 5,388,713 bp with a mean GC content of 66.7%. Gene predictions and annotations were performed with Rapid Annotation using Subsystem Technology (RAST) database (8) and NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (http://www.ncbi.nlm.nih.gov/genome/annotation_prok/process/). Based on RAST results, the draft genome contains 5318 coding sequences and 493 subsystems. There were 5421 genes, 342 pseudogenes and 1 CRISPR array annotated from NCBI PGAP. Three complete 5S rRNA, one partial 16S rRNA and two partial 23S rRNA encoding regions were predicted from RNAmmer 1.2 (9). In addition, 47 tRNAs were identified by tRNAscan-SE (10). The PHAST server (11) predicted five prophage regions with the sizes of 13 Kbp, 21.7 Kbp, 15.4 Kbp, 9.2 Kbp and 95.3 Kbp. Panseq tool (12) was used for analysing the core set and accessory genomic regions. It was found that some mobile elements, hypothetical proteins, uncharacterized conserved proteins, ABC transporter related proteins and some predicted transcriptional regulators of different families such as MerR, GntR and LacI are found as the novel regions in Paracoccus sp. MKU1 genome while comparing with other existing genomes of Paracoccus sp. As Paracoccus sp. is biochemically versatile genus, well known for its unique nitrate reduction properties and various metabolisms of utilizing variety of compounds, RAST functional annotations of Paracoccus sp. MKU1 showed that there are 99 genes involved in nitrogen metabolism, where 37 genes involved in denitrification; 206 genes involved in stress responses; 75 genes involved in resistance to antibiotics and toxic compounds including cobalt, zinc, cadmium, mercury and chromium (Figure 1). Analysis of genes for enzymes involved in DMF degradation pathways showed the presence of methylamine dehydrogenase and formamidase. Furthermore, there are 115 genes involved in degradation of various aromatic compounds, where 29 genes are involved in peripheral pathways for catabolism of aromatic compounds and 73 genes are involved in metabolism of central aromatic intermediates. The genome analysis showed the presence of genes involved in the degradation of various xenobiotics namely toluene, benzoate, p-hydroxy benzoate, aromatic amines, biphenyls, catechols and chloroaromatic compounds. In addition, pathways of salicylate and gentisate catabolism, protocatechuate and catechol branch of beta-ketoadipate pathways, homogentisate pathways known to be involved in aromatic hydrocarbons degradation are also found.
The overview of subsystem category coverage of Paracoccus sp. MKUI genome based on RAST server.
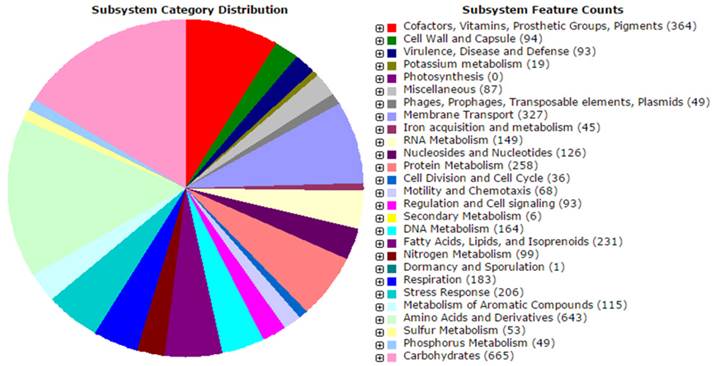
The draft genome of Paracoccus sp. MKU1 provide insights into the genes involved in the degradation of various xenobiotics, which can further open up the opportunities for potential exploitation of the versatile bacterium for bioremediation of xenobiotic compounds.
Nucleotide sequence accession number
The genome sequence of Paracoccus sp. MKU1 has been deposited at GenBank under the accession number LLWQ00000000. The version described in this paper is the first version. DNA sequence raw data has been uploaded in Sequence Read Archive (SRA) with the accession number SRX1601982. The Bioproject designation for this project is PRJNA298725.
Acknowledgements
Authors gratefully acknowledge SERB, Department of Science and Technology (DST), India for the financial support to BA [No.SR/FT/LS-29/2010] and DST-PURSE Program, Madurai Kamaraj University for the facilities. We are grateful to M/s Genotypic Technology Pvt. Ltd., Bengaluru, India, for the Illumina sequencing. We also thank Mr. R. Karthikeyan, School of Biological sciences, Madurai Kamaraj University for helping in Bioinformatics analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Baj J. Taxonomy of the genus Paracoccus. Acta Microbiol Pol. 2000;49:185-200
2. Urakami T, Araki H, Oyanagi H, Suzuki KI, Komagata K, Paracoccus aminophilus sp. nov. and Paracoccus aminovorans sp. nov, which utilize N,N-dimethylformamide. Int J Syst Bacteriol. 1990;40:287-291
3. Lin Q, Donghui W, Jianlong W. Biodegradation of pyridine by Paracoccus sp. KT-5 immobilized on bamboo-based activated carbon. Bioresour Technol. 2010;101:5229-5234
4. Zhang J, Zheng JW, Liang B, Wang CH, Ni YY, He J, Li SP. Biodegradation of chloroacetamide herbicides by Paracoccus sp. FLY-8 in vitro. J Agric Food Chem. 2011;59:4614-4621
5. Cai S, Cai T, Liu S, Yang Q, He J, Chen L, Hu J. Biodegradation of N-Methylpyrrolidone by Paracoccus sp. NMD-4 and its degradation pathway. Int Biodeter Biodegr. 2014;93:70-77
6. Nisha KN, Devi V, Varalakshmi P, Ashokkumar B. Biodegradation and utilization of dimethylformamide by biofilm forming Paracoccus sp. strains MKU1 and MKU2. Bioresour Technol. 2015;188:9-13
7. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina paired-end read merger. Bioinformatics. 2014;30:614-620
8. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75-89
9. Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100-3108
10. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955-964
11. Zhou Y, Liang Y, Lynch K, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347-W352
12. Laing C, Buchanan C, Taboada EN, Zhang Y, Kropinski A, Villegas A, Thomas JE Gannon VPJ. Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinformatics. 2010;11:461
Author contact
![]() Corresponding author: Balasubramaniem Ashokkumar. Tel: (91) 452-2459105; FAX: (91) 452-2459115; Email: rbashokkumarcom
Corresponding author: Balasubramaniem Ashokkumar. Tel: (91) 452-2459105; FAX: (91) 452-2459115; Email: rbashokkumarcom