Journal of Genomics
ISSN: 1839-9940
ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2014; 2:77-88. doi:10.7150/jgen.8123 This volume Cite
Review
Chromosome Imbalance as a Driver of Sex Disparity in Disease
Lara K. Abramowitz, Stéphanie Olivier-Van Stichelen, John A. Hanover ![]()
Laboratory of Cell and Molecular Biology, NIDDK, National Institutes of Health, Bethesda, MD 20892-0851, USA
Published 2014-4-1
Citation:
Abramowitz LK, Olivier-Van Stichelen S, Hanover JA. Chromosome Imbalance as a Driver of Sex Disparity in Disease. J Genomics 2014; 2:77-88. doi:10.7150/jgen.8123. https://www.jgenomics.com/v02p0077.htm
Other stylesAbstract
It has long been recognized that men and women exhibit different risks for diverse disorders ranging from metabolic to autoimmune diseases. However, the underlying causes of these disparities remain obscure. Analysis of patients with chromosomal abnormalities, including Turner syndrome (45X) and Klinefelter syndrome (47XXY), has highlighted the importance of X-linked gene dosage as a contributing factor for disease susceptibility. Escape from X-inactivation and X-linked imprinting can result in transcriptional differences between normal men and women as well as in patients with sex chromosome abnormalities. Animal models support a role for X-linked gene dosage in disease with O-linked N-acetylglucosamine transferase (OGT) emerging as a prime candidate for a pleiotropic effector. OGT encodes a highly regulated nutrient-sensing epigenetic modifier with established links to immunity, metabolism and development.
Keywords: dosage compensation, Turner syndrome, Klinefelter syndrome, imprinting, O-GlcNAcylation
Introduction
Men and women have different susceptibilities for a number of diseases including cognitive disorders, metabolic, cardiovascular and autoimmune disease. Although sex hormones account for some of the differences in predisposition to disease, it is unlikely to be the only causative factor. Intriguingly, observations of patients with sex chromosome abnormalities have implicated gene dosage of X-linked genes as a potential factor contributing to sex disparities in disease development. For example, a subset of Turner syndrome females have increased prevalence of cognitive disorders, visceral adiposity and atherogenic lipid profiles similar to men, whereas Klinefelter males have a predisposition to systemic lupus erythematosus (SLE) similar to women.
Turner syndrome (TS) is a chromosomal disorder caused by complete or partial loss of an X chromosome (45X karyotype), occurring in ~1/2500 live born females [1]. TS females exhibit many characteristic features, including short stature, gonadal dysgenesis, and organ abnormalities [2]. Interestingly, manifestations of many TS associated features are quite variable. For example, congenital cardiovascular and renal defects, as well as deficits in neuropsychological function are present only in smaller subsets of patients [3, 4]. The wide range of phenotypes exhibited by TS females is largely due to the fact that there are a number of genes on the X chromosome that escape X-inactivation and are normally biallelically expressed in 46XX females. In humans, ~15% of X-linked genes escape X-inactivation [5] and presumably, haploinsufficiency for one or more of these genes could cause the variable phenotypes associated with TS.
About 60-80% of TS females inherit their X chromosome from their mothers (XM) with the remaining having an X chromosome of paternal origin (XP) [6-9]. Intriguingly, characteristics such as cardiovascular disease, lipid metabolism, visceral adiposity and cognitive function have been associated with the parental origin of the single X chromosome in TS patients [6, 9-19]. In other words, a TS patient has an increased likelihood of these features depending on whether her X chromosome was inherited from her mother or father. These observations suggest that there are unique genes expressed exclusively from either the XM or XP, and led to the hypothesis of X-linked imprinted genes [11].
Klinefelter syndrome (KS) is the most common sex chromosome abnormality in men, occurring in about 1 in 660 newborn boys. These males have an extra X chromosome with the karyotype 47XXY. KS is greatly under diagnosed. Only ~25% of men with KS are diagnosed, with the mean age of diagnosis in the mid-30s. KS men are tall, have a gynecoid body habitus and are hypogonadal [20]. One intriguing feature of KS men is that, similar to women, they are 14 times more likely to develop SLE than normal XY males [21, 22].
In this review we present observations from TS and KS patients that highlight the importance of X-linked gene dosage in the susceptibility of disease. Here, we focus on the parent-of-origin effects in TS and consider how the mouse has been used to model these TS-like effects. Additionally, we discuss how activation of genes on the silenced X chromosome in females could lead to sex disparities in the predisposition to lupus. Finally, we summarize the emerging evidence that differential expression of the versatile epigenetic regulator O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) from the X chromosome may contribute to the gender disparities in disease susceptibility.
Sex chromosome dosage compensation
In mammals sex is determined by the presence or absence of a Y chromosome. Whereas females normally have two X chromosomes, males have an X and Y chromosome. Because of the imbalance in the number of chromosomes between males and females, mechanisms of dosage compensation have evolved. Dosage compensation is achieved in a two-fold manner in mammals; (1) by inactivation of one of the two X chromosomes in females [23] and (2) upregulation of X-linked genes to balance the expression levels between X-linked and autosomal genes [24-28].
Female cells undergo a process of random X-inactivation so that approximately half of the cells have an inactive XM and half an inactive XP. The process of X chromosome inactivation is largely mediated by the expression of the long non-coding RNA Xist [29, 30], which acts to recruit repressive complexes silencing one X chromosome [31]. DNA methylation is then necessary to maintain the silenced X chromosome [32, 33].
Despite being on the inactive X, many genes remain active. In humans, ~15% of genes on the X chromosome escape X-inactivation, whereas in mice only ~3-6% escape [5, 34, 35]. Some of these escape genes have a Y chromosome paralog, resulting in equal expression from both sexes, and homologous pairing during meiosis. These regions of homology between the sex chromosomes are known as pseudoautosomal regions (PAR) [36]. Other escape genes are expressed exclusively from the X chromosome, exhibiting higher expression in females [5, 34, 35]. Therefore, in TS patients, who only have one X chromosome, transcription of a number of genes is lower as compared to normal females. Conversely, in KS patients, who have two X and one Y chromosomes, transcription of genes in the PAR is greater than in normal males. Some of these genes have been directly correlated to specific disease characteristics. For example, haploinsufficiency of SHOX (short stature homeobox-containing gene) contributes to the short stature of TS females [37], however, overexpression of SHOX in KS males is associated with taller height [38].
In addition to X-inactivation, a second form of dosage compensation maintains a balance between X-linked and autosomal gene expression by doubling transcription from the active X chromosome [24-28]. Whereas little is known about the mechanisms coordinating upregulation of the X chromosome in mammals, this concept has been well documented in Drosophila. Similar to humans, Drosophila males and females are distinguished by their XY or XX karyotypes, respectively. In contrast to mammals, Drosophila males upregulate expression of their single X chromosome, and females maintain two active X chromosomes. In male somatic cells of Drosophila the male-specific lethal (MSL) complex, which is comprised of proteins and non-coding RNAs, is targeted to the X chromosome and is necessary for transcriptional upregulation. Importantly, the component males absent on the first (MOF) specifically acetylates histone H4 lysine 16 leading to opening of chromatin and increased expression [39]. Although further research is essential to elucidate the mechanisms of X-upregulation in mammals, insights from Drosophila can help in understanding the mammalian system. In fact, many of the MSL components have orthologues in humans, and MOF containing complexes are largely evolutionarily conserved [40].
X-linked imprinting
Genomic imprinting is an epigenetic phenomenon in which expression of a small subset of genes is dependent upon the parental origin of the chromosome [41]. Therefore, some genes will be expressed from the paternal allele, whereas others will be expressed from the maternal allele. One key feature of imprinted genes is the presence of differential DNA methylation that marks the parental origin of the allele [42]. Imprinting of autosomal genes has been well established and loss of imprinting of many genes causes human congenital diseases such as Beckwith-Wiedemann, Silver-Russell, Prader-Willie and Angelman syndromes [31, 43].
Due to the imbalance of X chromosomes between males and females, expression of X-linked imprinted genes would differ between the two sexes. While females have both a maternal and paternal X chromosome, males inherit only a maternal X. Therefore, if a gene were expressed exclusively from the paternal allele, it can only be expressed in females (figure 1A). However, if a gene were expressed from the maternal allele, expression levels would be higher in males than in females (figure 1B). The presence of imprinted X-linked genes could be one potential factor for differences in disease susceptibility between males and females.
One way to assess X-linked imprinting and its potential contributions to disease susceptibility is through analysis of TS patients. Phenotypic comparisons of TS females that are monosomic for XM or XP have given us insights into the potential role of X-linked imprinting in human disease and cognitive function. Additionally, the observed parent-of-origin effects in TS patients has led to the discovery of X-linked imprinted genes in mice. It should be noted, however, that due to small sample sizes and potential cryptic Y-chromosome mosaicisim of patients, there has been controversy as to whether phenotypes could be attributed specifically to parental origin of the X chromosome [44].
Figure 1
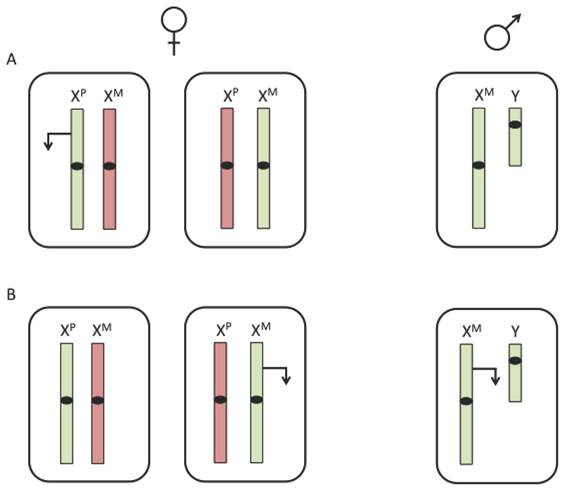
Expression of an X-linked imprinted gene. X-linked imprinting would result in differential expression between males and females. (A) A paternally expressed X-linked imprinted gene would only be expressed in about half of female cells and not expressed in male cells. (B) A maternally expressed X-linked imprinted gene would be expressed in all cells of a male and only half the cells of a female. The inactive X chromosome is represented in red, the active X chromosome is represented in green and transcription is represented by an arrow.
Parent-of-origin effects in Turner syndrome
Neuropsychological differences between XM and XP TS females
Impaired social cognition of TS females has been linked to the parental origin of the X chromosome. Using a questionnaire based approach to measure social cognition, it was reported that XM individuals scored significantly higher on measures of social cognitive dysfunction as compared to XP females. Similarly, normal males scored significantly higher than normal females, indicating poorer social cognition for both males and XM TS females [11]. Accordingly, a diagnosis of autism appears to be found more predominantly in XM versus XP TS patients [11, 12]. XM individuals also had a significantly lower verbal IQ and impaired behavioral inhibition [11]. However, a more recent study was unable to replicate the findings that XM TS females had poorer social cognition in a larger but younger cohort of patients [45]. Intriguingly, an additional study assessing verbal skills found that XP subjects rather than XM individuals had a significant decrease in verbal skills [13]. Hence, it still remains inconclusive whether or not there are X-linked imprinted genes involved in social cognition and verbal abilities.
Parent-of-origin effects have also been implicated in long-term memory. Verbal forgetting rate had been measured by assessing the ability of subjects to recall a story after delay. XM individuals were more likely to forget verbal material than XP females [15]. Interestingly, using the Rey figure copying task, in which a subject is asked to draw a figure from memory, XP individuals scored significantly lower than XM individuals [15]. Therefore, whereas XM females have deficits in verbal memory, XP individuals have deficits in visuospatial memory [15].
Studies of brain morphology comparing XM and XP TS females have suggested that imprinted X-linked genes are involved in brain development and anatomy [14, 16, 17]. These studies indicated that X-linked imprinted genes act to influence development in a region-specific manner. For example, XM females were reported to have a significant decrease in gray matter volume bilaterally in the caudate nuclei and thalamus, as well as decreased white matter volume in the temporal lobes [17]. Differences in volume have also been reported in the superior temporal gyrus in TS patients based on the parental origin of their X. Specifically, in XM females the right and left superior temporal gyrus were significantly larger than that of XP females [14, 16]. Similarly, a recent study has reported an increase in gray matter volume in the superior frontal and pericalcarine regions along with decreased white volume matter in the latter in XM TS females [14]. This same study also reported a bilateral increase in cortical thickness in the temporal and parietal lobes of XP TS individuals [14]. Although parent-of-origin effects are present in the brain morphology of TS patients, future research is necessary to determine if these morphological differences result in cognitive defects.
Eye disorders and hearing impairment in XM and XP females
Parent-of-origin effects have been described for eye disorders and sensorineural hearing loss, as both have significant increased prevalence in XP as compared to XM patients [9, 19]. The greater frequency of these disorders could potentially be due to differences in brain morphology between the two groups (as previously discussed) or due to monoallelic maternal expression of X-linked genes necessary for normal eye and hearing function.
Physical and structural anomalies in XM and XP females
Cardiac anomalies as well as neck webbing have also been associated with the parental origin of the X chromosome. XM females were reported to have a significant increase in cardiac abnormalities and neck webbing as compared to XP females [18]. The authors hypothesized that these features could be caused by abnormal development of the lymphatic system [18]. However, a more recent and larger study was unable to detect any significant differences in the presence of cardiac anomalies between XM and XP individuals [9]. Interestingly, Sagi et al. reported a significant increase in renal abnormalities in XM females as compared to XP females [9]. Similarly, Chu et al. observed more renal anomalies in XM than XP patients, though the differences were not statistically significant [18]. It therefore remains controversial as to whether some of the physical and structural anomalies associated with TS have a parent-of-origin component.
Adiposity and lipid metabolism in XM and XP individuals
In general, men have more visceral fat and atherogenic plasma lipids than women [46], making metabolic phenotypes a good candidate for X-imprinting effects. Young XM patients have been observed to be significantly overweight as compared to XP pediatric patients [9]. However, the XP pediatric patients in this study were reported to have higher total and low-density lipoprotein cholesterol levels [9]. A study of adult TS patients did not observe a difference in total body mass composed of fat, though the XM group had higher levels of abdominal fat and visceral fat as compared to the XP group [10]. Additionally, XM adults had a more atherogenic lipid profile, with higher triglyceride and low-density lipoprotein cholesterol levels [6, 10]. The conflicting observations of these studies could be due to the difference in ages of the subjects. Nevertheless, both studies support a role for imprinting of X-linked genes in metabolic regulation.
Mouse models of sex chromosome imbalance
The 39XO mouse
Unlike their human counterparts, mice monosomic for the X chromosome (referred to as 39XO) appear grossly normal and fertile. These differences in phenotypes between 39XO mice and TS females is most likely due to the fact that only ~3-6% of genes escape X-inactivation in the mouse, whereas about ~15% escape in humans. Therefore, many more genes exhibit decreased expression in TS patients than 39XO mice. Nevertheless, these mice possess some more subtle phenotypes similar to TS characteristics.
The 39XO mice were developmentally delayed early in gestation and had a reduction in body weight as well as a decreased germ cell population [47-50]. Additionally, 39XO mice had a higher frequency of hearing loss and decreased thyroid activity and body temperature [4, 51]. Studying these mice has some advantages that could help clarify many of the controversies in the human literature; firstly, mice possess a single X chromosome without the potential of Y chromosome mosaiscism. Secondly, the use of mice has the benefit of studying large, age-matched populations on the same genetic background.
Parent-of-origin effects in 39XO mice
39XO mice can be derived such that they specifically inherit a maternal or paternal X chromosome (39XMO and 39XPO, respectively). 39XPO mice are produced by mating a wild type male with a female containing a large inversion in her X chromosome (In(X)1h mutant) [4] (figure 2A). If crossing over within the large inversion occurs, the mother produces gametes without X chromosomes [52]. To generate 39XMO mice, wild type females are mated with Patchy fur (Paf) mutant males (figure 2B). Paf males have a mutation at the PAR, which results in nondisjunction during spermatogenesis, producing sperm without sex chromosomes [4, 53].
Figure 2
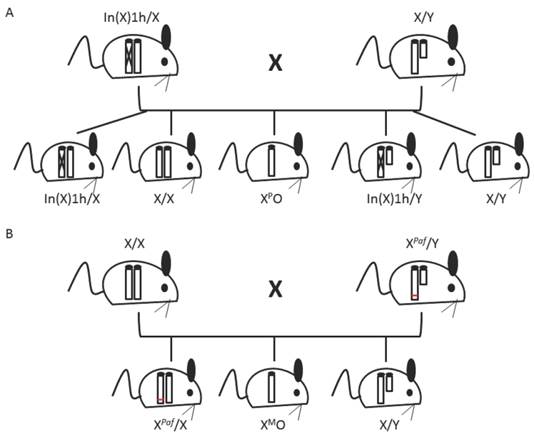
Mating schemes to derive monosomic X mice. (A) 39XPO mice are derived by mating an In(X)1h mutant female (In(X)1h/X) with a wild type male. The In(X)1h sex chromosome contains a large inversion (represented by an X in the chromosome). If crossing over during meiosis occurs within the inversion, gametes without an X chromosome will be produced. (B) 39XMO mice are derived by mating a wild type female (X/X) with a Paf mutant male (XPaf/Y). The Paf mutation (represented by a red line) is located at the pseudoautosomal region and causes nondisjunction during spermatogenesis, producing sperm without a sex chromosome. Genotypes of mice are indicated below.
Parent-of-origin effects have been described in the 39XO mouse model. 39XPO embryos had delayed postimplantation development, were smaller than 39XMO females and displayed poor development of the ectoplacental cone [54, 55]. These observations suggest that preferential expression of genes from the XM is necessary for normal embryogenesis. Interestingly, mice normally inactivate the paternal X chromosome in extraembryonic tissues [56]. Taken together, these data indicate that maternally expressed genes from the X chromosome could be required in extraembryonic tissues for normal development.
The 39XO model has also replicated some of the parental origin effects on cognitive function. Using a Y-maze based serial reversal learning paradigm, which tests the attentional and inhibitory processes required to switch from a pre-potent correct response to a previously incorrect response, 39XMO mice displayed significant deficits in reversal learning as compared to 39XPO mice [57]. These findings indicate that having only a maternal X chromosome led to difficulties in inhibiting response to a previously correct but now incorrect cue, and forming new associations with the previously incorrect but now correct cue [57].
Identification of X-linked imprinted genes in the mouse
Analysis of gene expression in 39XMO and 39XPO neonatal and embryonic mouse brains, has led to the identification of an X-linked imprinted gene cluster [57, 58]. Using Affymetrix microarrays Xlr3b, Xlr4b and Xlr4c were identified as being maternally expressed [57, 58]. The homologous region in the human, mapping to Xq28, contains a pseudogene with sequence homology to the Xlr3 gene family and has a high concentration of loci involved in neurodevelopmental pathologies [57, 58]. This pseudogene is most closely related to another human X-linked gene, FAM9B [57]. Although expression status of FAM9B in the brain has not been determined, deletions within this gene have been described in cases of autism and schizophrenia [59, 60].
Another X-linked imprinted gene has been identified in the mouse by comparing gene expression profiles of male and female blastocysts [61]. The X-linked gene, Rhox5, was determined to be expressed only in female blastocysts. Interestingly, imprinting of Rhox5 switches during development, with paternal expression observed from the 8-cell until the blastocyst stage and maternal expression observed at E7.5 [61]. Rhox5 does not have an obvious human orthologue.
The 'four core genotypes' mouse model of X-linked gene dosage and disease susceptibility
Another mouse model that uncouples sex hormones from sex chromosomes has correlated X-linked gene dosage with metabolic disorders associated with sex chromosome abnormalities. In mammals, sex determination is largely controlled by dosage-dependent nuclear accumulation of the Y-linked gene Sry, an architectural transcription factor that serves as a 'trigger for maleness' [62, 63]. This property of vertebrate sex determination has been exploited in the development of a model referred to as the four core genotypes mouse model. With this model, the Sry gene has been deleted from the Y chromosome and a functional Sry transgene was inserted onto an autosome, thus uncoupling testes determination from the Y chromosome [64, 65]. This allows generation of mice that can be XX or XY with female gonads and mice that are XX or XY with male gonads. The normal patterns of dosage compensation and inheritance of XMY are expected to occur independent of gonadal phenotype. The four core genotypes mouse model has been used to tease apart the influence of sex chromosomes from sex hormones for a variety of traits. Interestingly, mice with two X chromosomes had a greater body weight and more body fat than mice with an X and Y chromosome, regardless of gonadal sex. Moreover, when placed on a high fat diet these XX mice were faster to gain weight, and developed fatty liver and insulin resistance [66]. These results suggest that X-linked gene dosage plays a role in metabolic disease risk, irrespective of sex hormone levels. Accordingly, a high prevalence of type 2 diabetes has been reported in TS women, as well as KS men [67, 68].
OGT as a candidate X-linked gene associated with TS phenotypes
Aside from confirmed X-linked imprinted genes, transcriptional analysis of 39XO mice, as well as normal male and female mice, has uncovered a number of genes that exhibit both sex disparities in gene expression and dysregulation with X monosomy [69, 70]. Of interest, the gene encoding OGT, located in humans at Xq13.1 and in mice at XqD, is one such gene. Being located on the X chromosome, OGT is under the control of dosage compensation mechanisms [71, 72] (figure 3). However, Ogt has been identified as having decreased expression in 39XO mouse liver as well as sex dependent differences in expression in liver, adipocytes and placenta [69, 70, 73], suggesting that Ogt is either imprinted or escapes X-inactivation in these tissues.
Figure 3
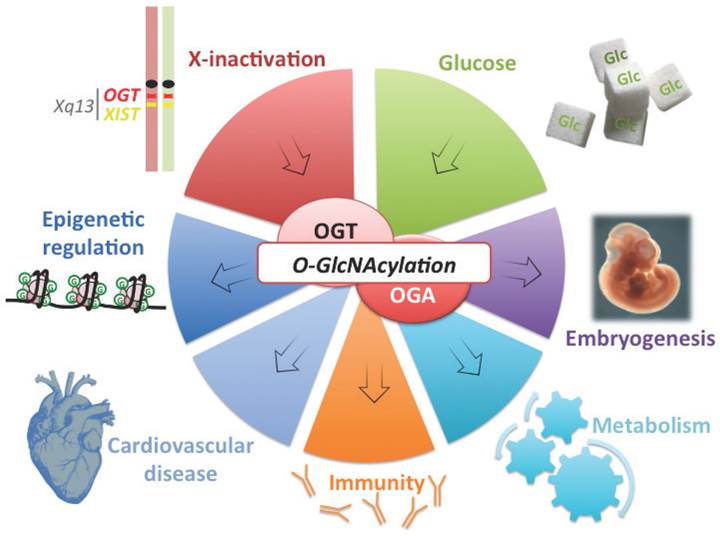
OGT is a good candidate connecting X-linked gene dosage to epigenetic regulation of metabolism and disease susceptibility. OGT is located on the X-chromosome, close to XIST and is subject to X-inactivation in mammalian females (silenced chromosome is depicted in red). Variation of OGT expression and the abundance of its substrate UDP-GlcNAc by glucose intake (Glc), modulate O-GlcNAc dynamism. This post-translational modification (represented by a green G) affects various processes from epigenetics to embryogenesis and immunity. O-GlcNAcylation is also involved in metabolism and cardiovascular defects, two of the major features of X-linked abnormalities.
OGT is a nutrient sensor that adds a single O-GlcNAc modification onto a variety of intracellular proteins. This modification is a key regulator of diverse cellular processes including, signal transduction, transcription and protein stability [74-76]. OGT utilizes the product of the hexosamine biosynthetic pathway, UDP-GlcNAc, as a sugar donor to add a GlcNAc moiety to serine and threonine residues of subsrates. This modification is removed by O-GlcNAcase (OGA or MGEA5). O-GlcNAcylation has been implicated in many human diseases such as type 2 diabetes, cancers, aging, cardiovascular and neurodegenerative diseases (figure 3) [74, 76]. Importantly, O-GlcNAcylation, and by extension OGT expression, plays a role in processes that contribute to parent-of-origin phentoypes in TS patients such as, gluconeogenesis, lipogenesis and cardiac metabolism [76-79].
OGT has recently emerged as a major epigenetic regulator. OGT has been found to complex with TET1,2,3 (regulators of DNA demethylation) [80-82], SIN3A and histone deacetylases (transcriptional repressors) [83], HCF-1 (transcriptional activator) [84, 85], MLL5 (histone methyltransferase) [86] and polycomb repressive complex 2 (PRC2) [87], in addition to being able to modify histones themselves [87-90]. Beyond interacting with epigenetic regulators, OGT modifies the C-terminal domain (CTD) of RNA polymerase II [91, 92]. Therefore, OGT is a general transcriptional regulator that could modulate diverse expression networks. Thus, potential dysregulation of OGT in TS patients could contribute to the variable phenotypes observed (figure 3).
Although OGT clearly acts as a key transcriptional regulator, how OGT itself is regulated to ensure proper gene dosage remains largely unknown. Because it is located close to Xist (figure 3), Ogt appears to be under tight transcriptional control in mice. Prior to widespread X-inactivation and upregulation of Xist, most X-linked genes are biallelically expressed in mouse embryonic stem (ES) cells. However, allelic analysis in mouse ES cells indicated that Ogt expression was skewed towards the paternal allele, suggesting that due to its close proximity to Xist, Ogt can be repressed prior to Xist upregulation [72].
Studies using Drosophila have also given us insights into potential regulatory mechanisms for human OGT. Evidence in Drosophila suggests that long introns can affect patterns of transcription and that Ogt expression is limited by splicing [93]. Similarly, the genes that encode OGT in mice and humans also contain unusually large introns. Moreover, many of the Drosophila dosage compensation components have orthologues in humans, and MOF containing complexes are evolutionarily conserved. One distinction between Drosophila MOF and human MOF complexes are the presence HCF-1 and OGT [40]. Thus, if human MOF has a role in upregulation of X-linked gene expression, OGT could be acting in a self-regulatory feedback loop.
Klinefelter syndrome and systemic lupus erythematosus
Women have an increased likelihood of developing autoimmune diseases, including SLE. In fact, about 90% of SLE patients are female [94]. One likely contributing factor is the hormonal differences between men and women. Estrogen exacerbates SLE in mouse models [95] and estrogen supplementation is associated with lupus flares in patients [96]. Nevertheless, sex hormones cannot be the only contributing factor for the sex differences in SLE as increased prevalence has also been observed in pre-pubertal girls and post-menopausal women [97, 98].
These observations have led to the hypothesis that a second X chromosome could predispose woman to SLE [22]. If this hypothesis were true, men with Klinefelter syndrome would be expected to develop SLE at a comparable rate to women. In fact, there is a 14-fold increase in the prevalence of SLE among men with Klinefelter syndrome as compared to men in the general population [21]. Therefore, males with an XXY karyotype are at a similar risk for developing SLE as females. Accumulating evidence suggests that activation of genes on the inactive X chromosome could contribute to the onset of SLE [22, 99, 100]. Thus, overexpression of X-linked genes could contribute to disease development particularly in women and KS men.
Aberrant DNA methylation patterns have been reported in CD4+ T cells from SLE patients [101-104], including hypomethylation of many X-linked genes [101]. It is therefore possible that demethylation on the silenced X chromosome could cause increased expression of X-linked genes contributing to autoimmunity in females and KS males. Accordingly, immune genes located on the X chromosome, including CD40LG, a T cell costimulatory molecule that plays an important role in T cell B cell interaction [105] and CXCR3, which encodes a chemokine receptor expressed in T cells [106], have been shown to be hypomethylated and overexpressed in women but not men with lupus [99, 100]. Of particular interest, OGT, which is required for T and B cell activation [107], was also identified as one of these hypomethylated/overexpressed genes [99, 100]. Tight regulation of dosage of these genes could be necessary for normal immune function. Thus, demethylation of these immune related genes on the inactive X chromosome could lead to increased expression, predisposing both women and KS men to SLE.
Conclusions and future directions
Observations from TS and KS patients highlight the importance of tight regulation of X-linked gene dosage and have given us insights into how dysregulation could contribute to sex disparities in disease development (figure 4). Parent-of-origin effects of the X chromosome in TS suggest the presence of genes whose expression depends on the parental origin of the allele. However, the confounding factors of cryptic mosaicism of the Y chromosome, and small populations of patients have made many of these studies difficult to replicate. Future studies with larger, aged-matched cohorts will be necessary to fully define parent-of-origin effects in TS. Nevertheless, these observations have led to the identification of X-linked imprinted genes in the mouse.
Further research focused on the identification of X-linked imprinted genes would not only help us tease apart the etiology of TS, but will also contribute to our knowledge of sex differences in the susceptibility to disease. Because males inherit only a maternal X, whereas females inherit both a maternal and paternal X, differences in gene expression from the X chromosome could contribute to sex specific disease risk. Interestingly, some of the features observed in XM patients, including cognitive dysfunction, increased visceral adiposity and a more atherogenic lipid profile are also more common among males.
Although 39XO mice are grossly normal and fertile, parent-of-origin effects on cognition and the identification of an X-linked imprinted cluster in the brain make the mouse a good model for exploring X-linked imprinting. Previous studies using the 39XO mouse have focused on the brain for identification of X-linked imprinted genes [57, 58], however, many imprinted genes are tissue-specifically imprinted [108] and therefore further transcriptional analysis in a variety of tissues is warranted. Furthermore, it will be important to ascertain the epigenetic mechanisms controlling X-linked imprinting. Whereas, allele-specific DNA methylation is a key feature for imprinting of autosomal genes [42], differential DNA methylation has not been identified at the Xlr imprinted cluster [109], suggesting a novel mechanism of regulation. Intriguingly, histone H1 depletion in the mouse resulted in upregulation of a number of imprinted genes, including Xlr3b and Rhox5 [110]. It is therefore possible that histone H1 is involved in the epigenetic mechanism regulating imprinting on the X chromosome.
As a master regulator of diverse cellular processes OGT has emerged as an X-linked gene requiring a specific dosage for normal function. Although recent evidence suggests OGT overexpression contributes to SLE, it remains unknown if OGT is dysregulated in TS individuals. Because of the overlap between TS characteristics and known roles of OGT, particularly in cardiovascular function, gluconeogenesis, lipogenesis and brain development, further examination of OGT expression in these individuals would be of interest. Moreover, elucidating mechanisms involved in regulation of OGT is necessary to understand how dosage is properly maintained and how changes could contribute to disease progression. Of particular interest, the gene encoding OGT has recently been shown to have differential expression between male and female placental tissue in both mice and humans [73]. As a nutrient sensor, this sex dependent disparity in expression poises OGT to differentially interpret environmental cues early in development with potential consequences manifesting later in life.
Historically, it has been difficult to obtain the quantities and numbers of appropriate patient samples required to statistically power the identification of candidate X-linked genes contributing to disease phenotypes. However, continuously evolving technologies put us on the precipice of understanding the potential role of X-linked gene dosage in human disease. The use of primary patient fibroblasts to generate induced pluripotent stem cells followed by high-throughput transcriptional analysis will provide a platform for identifying X-linked genes that contribute to disease risk. The molecular features of products encoded by these genes will help us better understand the role of X-linked gene dosage in sex-dependent disease risk and may provide novel therapeutic targets.
Figure 4
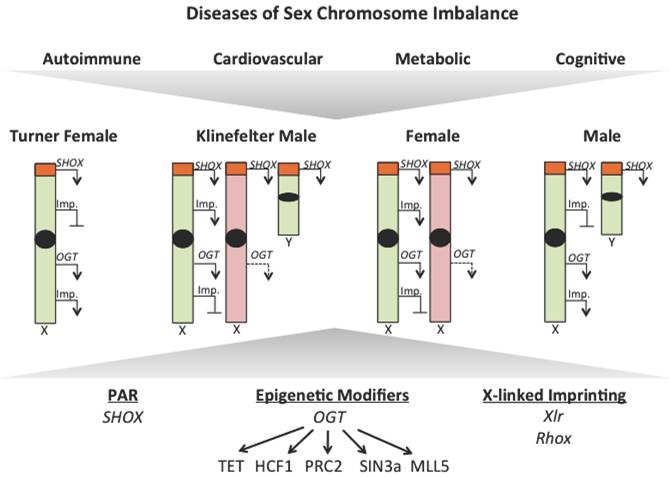
X-linked gene dosage contributes to sex disparities in disease. Observations from Turner and Klinefelter syndrome patients have uncovered a role for X-linked gene dosage in contributing to sex disparities in disease risk. Differences in expression of genes that escape X-inactivition, like those within the pseudoautosomal region, epigenetic modifiers, like OGT, and X-linked imprinted genes (Imp.) could all contribute to disease susceptibility. Active chromosomes are represented in green, inactive chromosomes are represented in red, transcription is represented with an arrow and loss of repression represented by a dotted arrow.
Abbreviations
TS: Turner syndrome; KS: Klinefelter syndrome; SLE: systemic lupus erythematosus; OGT: O-linked N-acetylglucosamine transferase; O-GlcNAc: O-linked N-acetylglucosamine; XM: maternal X chromosome; XP: paternal X chromosome; MOF: males absent on the first; MSL: male-specific lethal; SHOX: short stature homeobox-containing gene.
Acknowledgements
We thank members of the Hanover laboratory for helpful discussions. Our work is supported by National Institute of Diabetes and Digestive and Kidney Diseases intramural funds.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Stochholm K, Juul S, Juel K, Naeraa RW, Gravholt CH. Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab. 2006;91:3897-902 doi:10.1210/jc.2006-0558
2. Ranke MB, Saenger P. Turner's syndrome. The Lancet. 2001;358:309-14 doi:10.1016/s0140-6736(01)05487-3
3. Bondy CA, Cheng C. Monosomy for the X chromosome. Chromosome Research. 2009;17:649-58 doi:10.1007/s10577-009-9052-z
4. Lynn PMY, Davies W. The 39,XO mouse as a model for the neurobiology of Turner syndrome and sex-biased neuropsychiatric disorders. Behavioural Brain Research. 2007;179:173-82 doi:10.1016/j.bbr.2007.02.013
5. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400-4 doi:10.1038/nature03479
6. Alvarez-Nava F, Lanes R, Quintero JM, Miras M, Fideleff H, Mericq V. et al. Effect of the parental origin of the X-chromosome on the clinical features, associated complications, the two-year-response to growth hormone (rhGH) and the biochemical profile in patients with turner syndrome. Int J Pediatr Endocrinol. 2013;2013:10. doi:10.1186/1687-9856-2013-10
7. Uematsu A, Yorifuji T, Muroi J, Kawai M, Mamada M, Kaji M. et al. Parental origin of normal X chromosomes in Turner syndrome patients with various karyotypes: implications for the mechanism leading to generation of a 45,X karyotype. Am J Med Genet. 2002;111:134-9 doi:10.1002/ajmg.10506
8. Monroy N, Lopez M, Cervantes A, Garcia-Cruz D, Zafra G, Canun S. et al. Microsatellite analysis in Turner syndrome: parental origin of X chromosomes and possible mechanism of formation of abnormal chromosomes. Am J Med Genet. 2002;107:181-9
9. Sagi L, Zuckerman-Levin N, Gawlik A, Ghizzoni L, Buyukgebiz A, Rakover Y. et al. Clinical Significance of the Parental Origin of the X Chromosome in Turner Syndrome. Journal of Clinical Endocrinology & Metabolism. 2006;92:846-52 doi:10.1210/jc.2006-0158
10. Van PL, Bakalov VK, Zinn AR, Bondy CA. Maternal X chromosome, visceral adiposity, and lipid profile. JAMA. 2006;295:1373-4 doi:10.1001/jama.295.12.1373
11. Skuse DH, James RS, Bishop DV, Coppin B, Dalton P, Aamodt-Leeper G. et al. Evidence from Turner's syndrome of an imprinted X-linked locus affecting cognitive function. Nature. 1997;387:705-8 doi:10.1038/42706
12. Donnelly SL, Wolpert CM, Menold MM, Bass MP, Gilbert JR, Cuccaro ML. et al. Female with autistic disorder and monosomy X (Turner syndrome): parent-of-origin effect of the X chromosome. Am J Med Genet. 2000;96:312-6
13. Loesch DZ, Bui QM, Kelso W, Huggins RM, Slater H, Warne G. et al. Effect of Turner's syndrome and X-linked imprinting on cognitive status: analysis based on pedigree data. Brain and Development. 2005;27:494-503 doi:10.1016/j.braindev.2004.12.009
14. Lepage JF, Hong DS, Mazaika PK, Raman M, Sheau K, Marzelli MJ. et al. Genomic Imprinting Effects of the X Chromosome on Brain Morphology. Journal of Neuroscience. 2013;33:8567-74 doi:10.1523/jneurosci.5810-12.2013
15. Bishop DV, Canning E, Elgar K, Morris E, Jacobs PA, Skuse DH. Distinctive patterns of memory function in subgroups of females with Turner syndrome: evidence for imprinted loci on the X-chromosome affecting neurodevelopment. Neuropsychologia. 2000;38:712-21
16. Kesler SR, Blasey CM, Brown WE, Yankowitz J, Zeng SM, Bender BG. et al. Effects of X-monosomy and X-linked imprinting on superior temporal gyrus morphology in Turner syndrome. Biological Psychiatry. 2003;54:636-46 doi:10.1016/s0006-3223(03)00289-0
17. Cutter WJ, Daly EM, Robertson DMW, Chitnis XA, van Amelsvoort TAMJ, Simmons A. et al. Influence of X Chromosome and Hormones on Human Brain Development: A Magnetic Resonance Imaging and Proton Magnetic Resonance Spectroscopy Study of Turner Syndrome. Biological Psychiatry. 2006;59:273-83 doi:10.1016/j.biopsych.2005.06.026
18. Chu CE, Donaldson MD, Kelnar CJ, Smail PJ, Greene SA, Paterson WF. et al. Possible role of imprinting in the Turner phenotype. Journal of Medical Genetics. 1994;31:840-2 doi:10.1136/jmg.31.11.840
19. Hamelin CE. Genomic Imprinting in Turner Syndrome: Effects on Response to Growth Hormone and on Risk of Sensorineural Hearing Loss. Journal of Clinical Endocrinology & Metabolism. 2006;91:3002-10 doi:10.1210/jc.2006-0490
20. Groth KA, Skakkebaek A, Host C, Gravholt CH, Bojesen A. Clinical review: Klinefelter syndrome--a clinical update. J Clin Endocrinol Metab. 2013;98:20-30 doi:10.1210/jc.2012-2382
21. Scofield RH, Bruner GR, Namjou B, Kimberly RP, Ramsey-Goldman R, Petri M. et al. Klinefelter's syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 2008;58:2511-7 doi:10.1002/art.23701
22. Sawalha AH, Harley JB, Scofield RH. Autoimmunity and Klinefelter's syndrome: when men have two X chromosomes. J Autoimmun. 2009;33:31-4 doi:10.1016/j.jaut.2009.03.006
23. Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961;190:372-3
24. Deng X, Hiatt JB, Nguyen DK, Ercan S, Sturgill D, Hillier LW. et al. Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nature Genetics. 2011;43:1179-85 doi:10.1038/ng.948
25. Nguyen DK, Disteche CM. Dosage compensation of the active X chromosome in mammals. Nat Genet. 2006;38:47-53 doi:10.1038/ng1705
26. Gupta V, Parisi M, Sturgill D, Nuttall R, Doctolero M, Dudko OK. et al. Global analysis of X-chromosome dosage compensation. J Biol. 2006;5:3. doi:10.1186/jbiol30
27. Lin H, Halsall JA, Antczak P, O'Neill LP, Falciani F, Turner BM. Relative overexpression of X-linked genes in mouse embryonic stem cells is consistent with Ohno's hypothesis. Nat Genet. 2011;43:1169-70 author reply 71-2. doi:10.1038/ng.992
28. Kharchenko PV, Xi R, Park PJ. Evidence for dosage compensation between the X chromosome and autosomes in mammals. Nature Genetics. 2011;43:1167-9 doi:10.1038/ng.991
29. Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379:131-7 doi:10.1038/379131a0
30. Marahrens Y, Panning B, Dausman J, Strauss W, Jaenisch R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 1997;11:156-66
31. Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308-23 doi:10.1016/j.cell.2013.02.016
32. Sado T, Fenner MH, Tan SS, Tam P, Shioda T, Li E. X inactivation in the mouse embryo deficient for Dnmt1: distinct effect of hypomethylation on imprinted and random X inactivation. Dev Biol. 2000;225:294-303 doi:10.1006/dbio.2000.9823
33. Okamoto I, Heard E. Lessons from comparative analysis of X-chromosome inactivation in mammals. Chromosome Res. 2009;17:659-69 doi:10.1007/s10577-009-9057-7
34. Lopes AM, Arnold-Croop SE, Amorim A, Carrel L. Clustered transcripts that escape X inactivation at mouse XqD. Mamm Genome. 2011;22:572-82 doi:10.1007/s00335-011-9350-6
35. Yang F, Babak T, Shendure J, Disteche CM. Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res. 2010;20:614-22 doi:10.1101/gr.103200.109
36. Disteche CM. Dosage Compensation of the Sex Chromosomes. Annual Review of Genetics. 2012;46:537-60 doi:10.1146/annurev-genet-110711-155454
37. Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A. et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet. 1997;16:54-63 doi:10.1038/ng0597-54
38. Tuttelmann F, Gromoll J. Novel genetic aspects of Klinefelter's syndrome. Mol Hum Reprod. 2010;16:386-95 doi:10.1093/molehr/gaq019
39. Conrad T, Akhtar A. Dosage compensation in Drosophila melanogaster: epigenetic fine-tuning of chromosome-wide transcription. Nature Reviews Genetics. 2012;13:123-34 doi:10.1038/nrg3124
40. Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M. et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol Cell. 2006;21:811-23 doi:10.1016/j.molcel.2006.02.007
41. Bartolomei MS. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 2009;23:2124-33 doi:10.1101/gad.1841409
42. Abramowitz LK, Bartolomei MS. Genomic imprinting: recognition and marking of imprinted loci. Curr Opin Genet Dev. 2012;22:72-8 doi:10.1016/j.gde.2011.12.001
43. Thorvaldsen JL, Bartolomei MS. SnapShot: imprinted gene clusters. Cell. 2007;130:958. doi:10.1016/j.cell.2007.08.033
44. Henn W, Zang KD. Mosaicism in Turner's syndrome. Nature. 1997;390:569. doi:10.1038/37514
45. Lepage JF, Hong DS, Hallmayer J, Reiss AL. Genomic Imprinting Effects on Cognitive and Social Abilities in Prepubertal Girls with Turner Syndrome. Journal of Clinical Endocrinology & Metabolism. 2012;97:E460-E4 doi:10.1210/jc.2011-2916
46. Carr MC, Hokanson JE, Zambon A, Deeb SS, Barrett PH, Purnell JQ. et al. The contribution of intraabdominal fat to gender differences in hepatic lipase activity and low/high density lipoprotein heterogeneity. J Clin Endocrinol Metab. 2001;86:2831-7
47. Burgoyne PS, Baker TG. Perinatal oocyte loss in XO mice and its implications for the aetiology of gonadal dysgenesis in XO women. J Reprod Fertil. 1985;75:633-45
48. Burgoyne PS, Biggers JD. The consequences of X-dosage deficiency in the germ line: impaired development in vitro of preimplantation embryos from XO mice. Dev Biol. 1976;51:109-17
49. Burgoyne PS, Evans EP, Holland K. XO monosomy is associated with reduced birthweight and lowered weight gain in the mouse. J Reprod Fertil. 1983;68:381-5
50. Burgoyne PS, Tam PP, Evans EP. Retarded development of XO conceptuses during early pregnancy in the mouse. J Reprod Fertil. 1983;68:387-93
51. Hultcrantz M, Stenberg AE, Fransson A, Canlon B. Characterization of hearing in an X,0 'Turner mouse'. Hear Res. 2000;143:182-8
52. Evans EP, Phillips RJ. Inversion heterozygosity and the origin of XO daughters of Bpa/+female mice. Nature. 1975;256:40-1
53. Lane PW, Davisson MT. Patchy fur (Paf), a semidominant X-linked gene associated with a high level of X-Y nondisjunction in male mice. J Hered. 1990;81:43-50
54. Jamieson RV, Tan SS, Tam PP. Retarded postimplantation development of X0 mouse embryos: impact of the parental origin of the monosomic X chromosome. Dev Biol. 1998;201:13-25
55. Thornhill AR, Burgoyne PS. A paternally imprinted X chromosome retards the development of the early mouse embryo. Development. 1993;118:171-4
56. Schulz EG, Heard E. Role and control of X chromosome dosage in mammalian development. Current Opinion in Genetics & Development. 2013;23:109-15 doi:10.1016/j.gde.2013.01.008
57. Davies W, Isles A, Smith R, Karunadasa D, Burrmann D, Humby T. et al. Xlr3b is a new imprinted candidate for X-linked parent-of-origin effects on cognitive function in mice. Nat Genet. 2005;37:625-9 doi:10.1038/ng1577
58. Raefski AS, O'Neill MJ. Identification of a cluster of X-linked imprinted genes in mice. Nat Genet. 2005;37:620-4 doi:10.1038/ng1567
59. Thomas NS, Sharp AJ, Browne CE, Skuse D, Hardie C, Dennis NR. Xp deletions associated with autism in three females. Hum Genet. 1999;104:43-8
60. Milunsky J, Huang XL, Wyandt HE, Milunsky A. Schizophrenia susceptibility gene locus at Xp22.3. Clin Genet. 1999;55:455-60
61. Kobayashi S, Isotani A, Mise N, Yamamoto M, Fujihara Y, Kaseda K. et al. Comparison of gene expression in male and female mouse blastocysts revealed imprinting of the X-linked gene, Rhox5/Pem, at preimplantation stages. Curr Biol. 2006;16:166-72 doi:10.1016/j.cub.2005.11.071
62. Jager RJ, Anvret M, Hall K, Scherer G. A human XY female with a frame shift mutation in the candidate testis-determining gene SRY. Nature. 1990;348:452-4 doi:10.1038/348452a0
63. Hanover JA, Love DC, Prinz WA. Calmodulin-driven nuclear entry: trigger for sex determination and terminal differentiation. J Biol Chem. 2009;284:12593-7 doi:10.1074/jbc.R800076200
64. Arnold AP. Mouse models for evaluating sex chromosome effects that cause sex differences in non-gonadal tissues. J Neuroendocrinol. 2009;21:377-86 doi:10.1111/j.1365-2826.2009.01831.x
65. Link JC, Chen X, Arnold AP, Reue K. Metabolic impact of sex chromosomes. Adipocyte. 2013;2:74-9 doi:10.4161/adip.23320
66. Chen X, McClusky R, Chen J, Beaven SW, Tontonoz P, Arnold AP. et al. The number of x chromosomes causes sex differences in adiposity in mice. PLoS Genet. 2012;8:e1002709. doi:10.1371/journal.pgen.1002709
67. Bakalov VK, Cheng C, Zhou J, Bondy CA. X-chromosome gene dosage and the risk of diabetes in Turner syndrome. J Clin Endocrinol Metab. 2009;94:3289-96 doi:10.1210/jc.2009-0384
68. Tartaglia N, Davis S, Hench A, Nimishakavi S, Beauregard R, Reynolds A. et al. A new look at XXYY syndrome: medical and psychological features. Am J Med Genet A. 2008;146A:1509-22 doi:10.1002/ajmg.a.32366
69. Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L. et al. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006;16:995-1004 doi:10.1101/gr.5217506
70. Lopes AM, Burgoyne PS, Ojarikre A, Bauer J, Sargent CA, Amorim A. et al. Transcriptional changes in response to X chromosome dosage in the mouse: implications for X inactivation and the molecular basis of Turner Syndrome. BMC Genomics. 2010;11:82. doi:10.1186/1471-2164-11-82
71. Brown CJ, Lafreniere RG, Powers VE, Sebastio G, Ballabio A, Pettigrew AL. et al. Localization of the X inactivation centre on the human X chromosome in Xq13. Nature. 1991;349:82-4 doi:10.1038/349082a0
72. Lin H, Gupta V, VerMilyea MD, Falciani F, Lee JT, O'Neill LP. et al. Dosage Compensation in the Mouse Balances Up-Regulation and Silencing of X-Linked Genes. PLoS Biol. 2007;5:e326. doi:10.1371/journal.pbio.0050326
73. Howerton CL, Morgan CP, Fischer DB, Bale TL. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci U S A. 2013;110:5169-74 doi:10.1073/pnas.1300065110
74. Bond MR, Hanover JA. O-GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr. 2013;33:205-29 doi:10.1146/annurev-nutr-071812-161240
75. Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13:312-21 doi:10.1038/nrm3334
76. Ruan HB, Singh JP, Li MD, Wu J, Yang X. Cracking the O-GlcNAc code in metabolism. Trends Endocrinol Metab. 2013;24:301-9 doi:10.1016/j.tem.2013.02.002
77. Issad T, Kuo M. O-GlcNAc modification of transcription factors, glucose sensing and glucotoxicity. Trends Endocrinol Metab. 2008;19:380-9 doi:10.1016/j.tem.2008.09.001
78. Zachara NE. The roles of O-linked beta-N-acetylglucosamine in cardiovascular physiology and disease. Am J Physiol Heart Circ Physiol. 2012;302:H1905-18 doi:10.1152/ajpheart.00445.2011
79. Darley-Usmar VM, Ball LE, Chatham JC. Protein O-linked beta-N-acetylglucosamine: a novel effector of cardiomyocyte metabolism and function. J Mol Cell Cardiol. 2012;52:538-49 doi:10.1016/j.yjmcc.2011.08.009
80. Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561-4 doi:10.1038/nature11742
81. Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A. et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell. 2013;49:645-56 doi:10.1016/j.molcel.2012.12.019
82. Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N. et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645-55 doi:10.1038/emboj.2012.357
83. Yang X, Zhang F, Kudlow JE. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell. 2002;110:69-80
84. Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J. et al. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell. 2011;144:376-88 doi:10.1016/j.cell.2010.12.030
85. Daou S, Mashtalir N, Hammond-Martel I, Pak H, Yu H, Sui G. et al. Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proc Natl Acad Sci U S A. 2011;108:2747-52 doi:10.1073/pnas.1013822108
86. Fujiki R, Chikanishi T, Hashiba W, Ito H, Takada I, Roeder RG. et al. GlcNAcylation of a histone methyltransferase in retinoic-acid-induced granulopoiesis. Nature. 2009;459:455-9 doi:10.1038/nature07954
87. Myers SA, Panning B, Burlingame AL. Polycomb repressive complex 2 is necessary for the normal site-specific O-GlcNAc distribution in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2011;108:9490-5 doi:10.1073/pnas.1019289108
88. Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S. et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557-60 doi:10.1038/nature10656
89. Sakabe K, Hart GW. O-GlcNAc transferase regulates mitotic chromatin dynamics. J Biol Chem. 2010;285:34460-8 doi:10.1074/jbc.M110.158170
90. Fong JJ, Nguyen BL, Bridger R, Medrano EE, Wells L, Pan S. et al. beta-N-Acetylglucosamine (O-GlcNAc) is a novel regulator of mitosis-specific phosphorylations on histone H3. J Biol Chem. 2012;287:12195-203 doi:10.1074/jbc.M111.315804
91. Comer FI, Hart GW. Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry. 2001;40:7845-52
92. Ranuncolo SM, Ghosh S, Hanover JA, Hart GW, Lewis BA. Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo. The Journal of biological chemistry. 2012;287:23549-61 doi:10.1074/jbc.M111.330910
93. Ashton-Beaucage D, Udell CM, Lavoie H, Baril C, Lefrancois M, Chagnon P. et al. The exon junction complex controls the splicing of MAPK and other long intron-containing transcripts in Drosophila. Cell. 2010;143:251-62 doi:10.1016/j.cell.2010.09.014
94. Stewart JJ. The female X-inactivation mosaic in systemic lupus erythematosus. Immunol Today. 1998;19:352-7
95. Roubinian JR, Papoian R, Talal N. Androgenic hormones modulate autoantibody responses and improve survival in murine lupus. J Clin Invest. 1977;59:1066-70 doi:10.1172/jci108729
96. Buyon JP, Petri MA, Kim MY, Kalunian KC, Grossman J, Hahn BH. et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med. 2005;142:953-62
97. Lockshin MD. Biology of the sex and age distribution of systemic lupus erythematosus. Arthritis Rheum. 2007;57:608-11 doi:10.1002/art.22676
98. Lockshin MD. Sex ratio and rheumatic disease: excerpts from an Institute of Medicine report. Lupus. 2002;11:662-6
99. Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol. 2007;179:6352-8
100. Hewagama A, Gorelik G, Patel D, Liyanarachchi P, McCune WJ, Somers E. et al. Overexpression of X-linked genes in T cells from women with lupus. J Autoimmun. 2013;41:60-71 doi:10.1016/j.jaut.2012.12.006
101. Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J. et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet. 2013;9:e1003678. doi:10.1371/journal.pgen.1003678
102. Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J. et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20:170-9 doi:10.1101/gr.100289.109
103. Jeffries MA, Dozmorov M, Tang Y, Merrill JT, Wren JD, Sawalha AH. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics. 2011;6:593-601
104. Zhang Y, Zhao M, Sawalha AH, Richardson B, Lu Q. Impaired DNA methylation and its mechanisms in CD4(+)T cells of systemic lupus erythematosus. J Autoimmun. 2013;41:92-9 doi:10.1016/j.jaut.2013.01.005
105. Lederman S, Yellin MJ, Cleary AM, Pernis A, Inghirami G, Cohn LE. et al. T-BAM/CD40-L on helper T lymphocytes augments lymphokine-induced B cell Ig isotype switch recombination and rescues B cells from programmed cell death. J Immunol. 1994;152:2163-71
106. Enghard P, Humrich JY, Rudolph B, Rosenberger S, Biesen R, Kuhn A. et al. CXCR3+CD4+ T cells are enriched in inflamed kidneys and urine and provide a new biomarker for acute nephritis flares in systemic lupus erythematosus patients. Arthritis Rheum. 2009;60:199-206 doi:10.1002/art.24136
107. Golks A, Tran TT, Goetschy JF, Guerini D. Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. EMBO J. 2007;26:4368-79 doi:10.1038/sj.emboj.7601845
108. Prickett AR, Oakey RJ. A survey of tissue-specific genomic imprinting in mammals. Mol Genet Genomics. 2012;287:621-30 doi:10.1007/s00438-012-0708-6
109. Qureshi S, Murphy M, Kasowitz S, Foley R, O'Neill M. Identification of a novel imprinting mechanism at the X-linked imprinted locus, X-linked Lymphocyte Regulated 3/4 (Xlr3/4). Epigenetics Chromatin. 2013 6
110. Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE. et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell. 2005;123:1199-212 doi:10.1016/j.cell.2005.10.028
Author contact
![]() Corresponding author: John A. Hanover. Tel: 301-496-0943; Fax: 301-496-9431; Email: johnhniddk.nih.gov
Corresponding author: John A. Hanover. Tel: 301-496-0943; Fax: 301-496-9431; Email: johnhniddk.nih.gov
Citation styles
APA
Abramowitz, L.K., Olivier-Van Stichelen, S., Hanover, J.A. (2014). Chromosome Imbalance as a Driver of Sex Disparity in Disease. Journal of Genomics, 2, 77-88. https://doi.org/10.7150/jgen.8123.
ACS
Abramowitz, L.K.; Olivier-Van Stichelen, S.; Hanover, J.A. Chromosome Imbalance as a Driver of Sex Disparity in Disease. J. Genomics 2014, 2, 77-88. DOI: 10.7150/jgen.8123.
NLM
Abramowitz LK, Olivier-Van Stichelen S, Hanover JA. Chromosome Imbalance as a Driver of Sex Disparity in Disease. J Genomics 2014; 2:77-88. doi:10.7150/jgen.8123. https://www.jgenomics.com/v02p0077.htm
CSE
Abramowitz LK, Olivier-Van Stichelen S, Hanover JA. 2014. Chromosome Imbalance as a Driver of Sex Disparity in Disease. J Genomics. 2:77-88.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.